当涉及到解剖细胞的调节回路是如何连接时,一些研究人员转向他们的移液管。Emily Miraldi转向键盘。
作为俄亥俄州辛辛那提儿童医院的计算和系统生物学家,Miraldi用数学来理解是什么使细胞系统运转,并预测它们如何对环境做出反应。作为博士后,她与纽约市纽约大学的计算生物学家Richard Bonneau和免疫学家Dan Littman一起工作。2006年,Bonneau和他的同事们建立了一个名为Inferelator的计算建模工具1它使用基因表达数据来推断被称为转录因子的dna结合蛋白如何控制特定基因的表达。研究人员可以使用由此产生的网络图来跟踪细胞中的信息流,识别(也许还可以逆向工程)控制关键过程的调节器。
但是推断这些电路的结构是复杂的。即使是最简单的基因表达数据也可以用多种网络架构来解释,而看似直接的相互作用可能并非如此。转录因子通常协同工作,由酶修饰,可以在距离目标基因数万或数十万个DNA碱基的地方起作用。虽然在人类基因组中已经鉴定出了大约1600种转录因子,但关于它们与DNA结合的确切序列(或“母题”)的信息仍然缺乏。此外,细胞中的基因组DNA被包裹在一种叫做染色质的复合物中,它可以阻止转录因子的结合。
为了解决其中一些问题,Bonneau的团队引入了另一种实验数据来改进推断器。他们使用了一种技术的信息,该技术揭示了基因组中染色质的哪些区域是未包装的,可用于转录因子结合。这种方法被称为atac -测序法,用于高通量测序转座酶可达染色质。通过重新配置软件来使用这些数据,研究小组能够找出哪些基因串联地改变了表达,以及哪些转录因子dna结合基序可以影响这种表达。
Bonneau现在在加州南旧金山的基因泰克研究和早期开发公司工作,他称之为“力作”研究2Miraldi和她的同事们使用这种更新的推断器来追踪一类被称为17型t辅助细胞的白细胞中包含数千种转录因子的网络。他们发现这些细胞中的转录因子STAT3和FOXB1是炎症性肠病相关基因的关键调节因子。
Miraldi说:“这篇论文是我们第一次能够验证,如果你只从RNA-seq和ATAC-seq[数据]开始,你可以得到相对于单独的基因表达数据更准确的基因调控网络。”
今天,inferator只是一个快速增长的用于基因调节网络(GRN)推断的软件工具集合之一,无论是在群体水平还是单个细胞水平。这些方法可能仅仅依赖于基因表达数据,但也有一些利用了其他数据类型,或者模拟了调控网络的系统性破坏。其他人正在帮助梳理出指导转录因子活性的序列。Miraldi说,如果你想预测细胞的行为,“你需要了解它们是如何连接的”。
推论问题
研究人员可以通过实验梳理出调节网络。例如,使用染色质免疫沉淀(使用抗体来确定转录因子何时何地与DNA结合)和基因表达分析等方法,研究人员可以将转录因子结合与基因表达联系起来,并确定它们作用的DNA区域。从那里,他们可以建立网络来解释数据。但这些方法都是劳动密集型的,而且可能需要尚未制造或质量较差的抗体。他们倾向于一次只吃一种蛋白质。而我们感兴趣的细胞类型可能无法在实验室中获得或不切实际。GRN推理允许研究人员通过挖掘基因表达数据来计算推导这些网络来规避这些问题。由此产生的网络可以为实验设计提供信息,而实验设计反过来又可以改进计算模型。
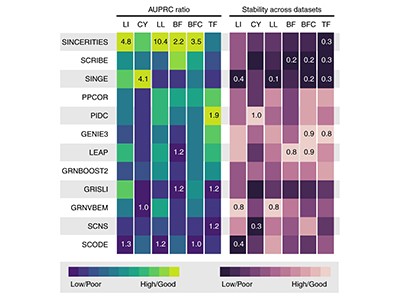
从单细胞转录组数据进行基因调控网络推断的基准算法
GRN推断最简单的方法依赖于相关性——成对基因表达同步上升和下降的趋势。亚特兰大佐治亚理工学院(Georgia Institute of Technology)的计算科学家张修维(Xiuwei Zhang)说:“如果我看到这两个基因在细胞间总是一起上升和下降,它们总是相关的,那么它们之间很有可能存在调控关系。”她已经建立了自己的grn推断工具。
比利时鲁汶天主教大学(KU Leuven)的博士生Seppe De Winter帮助开发了另一个grn推断工具,称为SCENIC+,利用了机器学习。或者,研究人员可以将grn简化为数学方程。今年1月,乔安娜·汉兹利克(Joanna Handzlik)使用了一种称为基因回路的建模方法——一种耦合微分方程系统,每个微分方程描述一个基因——来推导参与血细胞成熟的十几个转录因子和靶基因之间的调节关系3..
由于这样的模型需要大量的计算,研究人员倾向于通过加入更少的蛋白质或将它们简化为布尔系统,其中每个相互作用要么是打开的,要么是关闭的。相反,Handzlik在这个问题上投入了计算能力。她在大学的高性能计算集群上并行运行了100个计算机处理核心数天,求解了数千万次方程,直到她得到了一组反映实验数据的模型参数。然后,Handzlik模拟了如果她消除或减少两个转录因子(PU.1和GATA1)中的任何一个的表达会发生什么。她说:“我们看到,值得注意的是,这个模型实际上与实验预期一致。”
“啊哈”的时刻
Aviv Regev是单细胞生物学的先驱,现在是基因泰克研究和早期开发的执行副总裁,她的大部分职业生涯都在追求grn。她说,促使她的团队设计更微妙的方法来处理和分析单细胞的动机之一,“源于这个主题对我来说是多么重要”。
她说,假设你扰乱了一群细胞中的一个基因。通过观察哪些基因受到影响,你可以建立一个调节回路模型。但为了证实你的假设,你可能需要破坏几十个甚至数百个其他基因。她说,这很快就变得不切实际了——但在单细胞水平上不是这样,在单细胞水平上,每个细胞都是自己的数据集。“我们认为,在单细胞基因组学方面,我们将能够做一些我们根本无法大规模做的事情。”
Regev和她的团队应用单细胞方法和新的计算方法,研究了来自骨髓的18个特化免疫细胞(称为树突状细胞)样本如何对细菌细胞壁的一种成分做出反应。他们说,这18个细胞实际上代表了两个种群。专注于更大的亚群,他们发现,尽管所有的亚群都同时受到细菌分子的刺激,但并不是所有的亚群都有相同程度的反应。利用细胞之间的微妙差异,研究小组推导出一个简单的相关电路,将转录因子STAT2和IRF7标记为抗病毒活性的“主调节因子”4.她说:“你可以从单细胞之间的这种变异中做很多事情。”
对于威斯康星大学麦迪逊分校的计算生物学家安东尼·吉特来说,雷格夫的工作代表了一个“啊哈”时刻。他发现,通过检查每个单细胞的轮廓,寻找它们在细胞分化路径上的相对位置的线索,就有可能在“伪时间”中按时间顺序将它们组织起来。
“伪时间可以让你对细胞排序,这样你就可以看到是什么导致了前面的效应,”Gitter说。它试图“通过使用一个细胞相对于其他细胞的表达测量来估计每个细胞的时间点”。然后,研究人员可以使用这些伪时间估计来构建grn。

CellOracle软件可以可视化改变细胞身份的基因调节网络。在自然状态下(左图),小鼠血细胞分化为红细胞(左下)或白细胞(左上);右边的面板显示了当关键转录因子被删除时的相同过程。图源:Kenji Kamimoto/Samantha Morris实验室
Gitter的团队基于这个想法创建了一个名为SINGE的工具5并将其应用于小鼠胚胎干细胞发育成内胚层细胞。他说,这种方法奏效了,但效果并不理想。“如果你唯一要研究的数据是基因表达,那么你对基因调控的了解似乎仍然存在一些根本的限制。”哈佛大学博德研究所(Broad Institute of Harvard)和麻省理工学院(MIT)基因调控观察站(Gene Regulation Observatory)联合主任杰森•布恩罗斯特罗(Jason Buenrostro)说,问题在于,基因表达数据本身无法充分“限制”能够解释这些数据的可能网络的数量。例如,两个相关的基因可以由同一个转录因子调控,也可以由第三个不同的转录因子调控的两个不同的基因调控。
在2020年的一项研究中,布莱克斯堡弗吉尼亚理工大学的计算机科学家T. M. Murali和他的团队描述了一种名为BEELINE的计算管道,他们用它来测试基于单细胞RNA测序的十几种grn推断方法,对比黄金标准和合成数据集6.穆拉利说:“大多数方法在推理方面做得相对较差,”至少在推断相互作用方面——表现和随机预测器差不多。他说,解决办法是加入额外的数据。
例如,Buenrostro的团队开发了一种名为FigR的计算框架。它使用来自单细胞RNA测序和ATAC-seq的数据,将转录因子及其靶基因的表达与蛋白质结合基序的鉴定和染色质可及性的数据整合在一起。“当我们这样做时,我们开始非常清楚地看到,许多与我们最喜欢的基因共表达的转录因子实际上并没有在我们最喜欢的基因上富集序列。”他说,这意味着没有转录因子结合和调节基因的地方,所以“它们从分析中被移除”。“我们还看到许多序列被富集,但转录因子甚至没有表达。”
基因调控网络的可复制推断、分析和出版的在线笔记本资源
最新版本的Inferelator还使用了单单元ATAC-seq数据。但它通过考虑转录因子活性进一步限制了这一信息。
“在你从测序数据中观察到的时候,转录因子的表达水平并不能表明它在做什么,”领导开发更新版本的Claudia Skok Gibbs解释说7.这是因为它们中的一些会与伴侣一起作用,或者必须经过化学修饰才能起作用。或者,它们的绑定位点可能无法进行绑定。Inferelator 3.0着眼于靶基因的表达水平以及转录因子基序数据库和基因组中潜在结合位点的染色质可及性。这意味着它可以确定哪些转录因子可用于刺激或抑制给定细胞类型中的靶基因。这些活动分数随后被输入三种网络构建算法中的一种。
但是对于计算模型来说,它们包含的变量越多,它们就越好,Bonneau说。在许多情况下,性能的提高归结于噪声。为了平衡这些相互竞争的力量,他说,软件对模型中的每个蛋白质都给予“惩罚”——除非该蛋白质似乎在感兴趣的基因上活跃。“如果这种转录因子在靶基因附近有一个结合位点,并且在ATAC-seq数据中对该细胞类型也显示为开放的,我们认为它不必付出那么大的代价。”
Skok Gibbs使用Inferelator 3.0来识别大脑细胞中称为髓质神经元的调节因子果蝇果蝇8.这些神经元有几种形式,通过改变单个基因的表达可以将一种神经元转换为另一种。她说:“我能够证明我可以找到特定的转录因子,以及它针对的是什么基因,这是造成这种情况的原因。”
遗传变异的数据也可以用于GRN推断。在过去的十年里,马萨诸塞州波士顿哈佛大学公共卫生学院的网络生物学家John Quackenbush和他的团队创建了一个虚拟的算法“动物园”,命名为PANDA、LIONESS和CONDOR。这些方法利用了一种称为消息传递的机器学习策略,以及转录因子在基因组中可能结合的位置的知识,来猜测并优化GRN。该团队最近的迭代,EGRET,利用遗传变异的信息来定制grn,以适应特定的个体和细胞类型。这主要是通过考虑被称为多态性的序列变异如何影响转录因子结合来实现的9.
由此产生的网络可以揭示基因组非编码部分的变异如何导致疾病。在对119名来自西非约鲁巴人后裔的分析中,Quackenbush和他的同事们表明,与冠状动脉疾病相关的多态性主要影响心脏细胞中的grn,而与自身免疫性疾病相关的多态性则影响免疫细胞9.“在我们观察的最相关的细胞类型中,我们看到了我们预测的疾病相关转录因子基因调控的中断,”该研究的合著者黛博拉·威希尔说。
淘汰计划
2016年,雷格夫和剑桥麻省理工学院的细胞生物学家乔纳森·韦斯曼(Jonathan Weissman)以及他们的同事撰写了两项研究10,11描述了基于基因编辑技术CRISPR的混合筛选方法Perturb-seq。扰动-seq允许研究人员使用单细胞rna测序作为读数来减少或敲除选定的基因。Weissman说,以前的crispr筛选方法要么倾向于使用基因报告器,要么倾向于观察特定的表型。但是很多生物将在这种策略的雷达下飞行。韦斯曼说:“Aviv和我各自想出了这个主意,通过RNA测序,你基本上可以一次性观察所有的转录反应。”“这将给你更多的信息,并引导你了解基因的真正潜在功能是什么。”
在一项研究中10,研究人员使用Perturb-seq分析了24个转录因子对参与刺激骨髓来源的树突状细胞的基因的影响。另一方面11他们的目标是与一种称为未折叠蛋白反应的细胞应激途径相关的基因。从那时起,Regev将该方法移植到动物身上,并将其与蛋白质定量方法相结合,称为Perturb-CITE-seq。与此同时,韦斯曼的团队已经对Perturb-seq全基因组进行了检测,在超过250万个细胞中敲除了近1万个人类基因12.“所以现在你已经用各种可能的方式动摇了细胞,你在问,它是如何反应的?”斯曼说。

NatureTech中心
或者,研究人员可以扰乱遗传网络在网上.密苏里州圣路易斯华盛顿大学医学院Samantha Morris实验室的干细胞和发育生物学家Kenji Kamimoto创建了CellOracle,这是一种混合单细胞rna测序和ATAC-seq数据的软件工具,首先推断出GRN,然后破坏它。通过检查由此产生的细胞命运图的变化,研究人员可以可视化转录因子中断如何改变细胞群。
Kamimoto应用CellOracle系统地研究了可以对结缔组织细胞进行重编程,从而形成其他类型细胞的蛋白质,确定了可以大大提高这种转变效率的因素13.莫里斯说,至少有5项同行评议研究和13份预印本也使用了该工具。在一个14鲁汶大学的生物医学工程师Tim Herpelinck和他的同事使用CellOracle模拟了骨发育过程中转录因子Sox9的损失。“基因敲除实验需要大量的时间,特别是如果你想做的话在活的有机体内Herpelinck说。他补充说,Sox9基因的缺失对发育中的胚胎是致命的,因此这种分析尤其困难。
验证,验证,再验证
为了正确利用ATAC-seq数据,研究人员必须知道转录因子结合位点在哪里。Miraldi说,通常情况下,研究人员发现他们使用的是一种本质上的文本匹配算法。但在7月,她和她的团队描述了另一种选择:使用深度神经网络在ATAC-seq数据中找到这些位点。Miraldi表示,研究人员可以使用这种名为maxATAC的算法来模拟罕见细胞中的染色质免疫沉淀和DNA测序,对这些细胞进行这样的实验是不切实际的,包括在患者的样本中。Miraldi的团队使用maxATAC将转录因子MYB和FOXP1与一种称为特应性皮炎的常见自身免疫性疾病联系起来15.
Miraldi说,在寻找结合位点方面,该算法比传统的转录因子-基序扫描方法要好四倍。这应该“直接转化为基因调控网络推理的改进,因为你在识别转录因子结合位点方面更加准确”。但它不能找到所有的东西:maxATAC只包含了近1600种已确定的人类转录因子中的127种。
为了帮助缩小差距,研究人员可以再次转向深度学习。2021年,加利福尼亚州斯坦福大学的计算生物学家Anshul Kundaje和密苏里州堪萨斯城斯托尔斯医学研究所的Julia Zeitlinger描述了一种名为BPNet的卷积神经网络。它使用一种称为ChIP-nexus的染色质免疫沉淀数据,通过单核苷酸分辨率精确地了解转录因子与哪些DNA序列结合——至少在研究人员拥有数据的细胞中是这样16.这对研究人员将这种方法应用于用于制造诱导多能干细胞的四种转录因子——Oct4、Sox2、Klf4和Nanog——并在这些蛋白质如何与干细胞中的DNA结合方面发现了意想不到的微妙之处。例如,Nanog通常与Sox2结合,但前提是蛋白质的结合位点间隔10.5个碱基,这个距离与DNA螺旋的周期性相对应。昆达杰说:“即使对四种非常充分研究的多能性因素,我们也发现了新的协作模式。”
无论你选择哪种GRN方法,最终它都只是一个假设。像所有的生物信息学问题一样,GRN推理总是会返回一个答案。但莫里斯说,要确定这个答案是否有意义,你需要“验证、验证、验证”。
雷格夫说,随着方法变得越来越复杂,挑战就变成了规模问题:在某种程度上,不可能测试每个变量和组合。“世界上没有足够的细胞,”她说。但是,她指出,有可能设计出足够有效的实验,让研究人员无需实际测试就能预测其他实验结果。
使用Perturb-seq的另一种方法提供了一种解决方案,即观察同一单元中多个扰动的影响。在他们2016年的论文中10例如,Regev和她的团队发现,一些细胞每个细胞接收了多达三个crispr靶向rna。将这些细胞与只接受了一到两个靶向rna的细胞进行比较,他们发现在某些情况下,影响是协同的,这表明调控相互作用。她说,这种组合研究是“前沿——这是该领域的发展方向。”
一旦研究人员能够弄清楚细胞的线路,他们就可以修补它来改造细胞或修复它们。布恩罗斯特拉说:“可以说,这是生物学中最重要的问题。”
 从单细胞转录组数据进行基因调控网络推断的基准算法
从单细胞转录组数据进行基因调控网络推断的基准算法 基因调控网络的可复制推断、分析和出版的在线笔记本资源
基因调控网络的可复制推断、分析和出版的在线笔记本资源 用于单细胞基因调控网络分析的可伸缩的SCENIC工作流程
用于单细胞基因调控网络分析的可伸缩的SCENIC工作流程 NatureTech中心
NatureTech中心