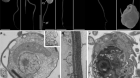
研究人员已经开发了一系列技术来解剖单个细胞的生物学,比如这些皮肤细胞。信贷:Vshivkova /上面
单细胞生物学在过去的十年里飞速发展。2015年至2021年期间,PubMed生物医学文献数据库中使用“单细胞”一词的研究增加了两倍多,这主要是由分离单细胞及其分子含量的技术创新推动的。
研究人员使用这些方法和许多其他方法以不同的方式探测单个细胞,从描述它们的基因表达到记录它们的表观遗传状态,转录因子活性和细胞间的通信。
密苏里州圣路易斯华盛顿大学的发育生物学家萨曼莎·莫里斯(Samantha Morris)说:“单细胞生物学已经真正向更广泛的受众开放了。”
但是,尽管湿实验室技术和分析方法的普及扩大了单细胞研究的范围,但它也使试图选择最佳方法的研究人员混淆了水。在线单细胞RNA工具目录,scRNA-tools,列出了近1400个软件包,可以将单细胞数据转化为科学见解。研究人员应该如何选择呢?
奥斯陆大学的生物信息学家Geir Kjetil Sandve说:“如果你要做任何类型的分析,你都面临着一个选择,而作为一名研究人员,你被期望做出合理的选择。”
德国海德堡大学的计算生物学家Julio Saez-Rodriguez说,科学家做出的选择很重要。“即使是很小的变化也会导致结果的巨大差异,”他说。
海星事业:在单细胞中寻找RNA模式
该领域分析丰富的解决方案是基准测试:对多种方法进行测试(理想情况下由中立方进行),通常应用于几种数据集,以确定哪种方法最适合不同的目的。自从单细胞研究出现以来,科学家们已经对湿实验室分析和分析算法进行了数十次比较,这可以指导研究人员为自己的工作选择方法。尽管一些研究人员表示,这些努力没有得到资助机构的重视,但新的举措正在为进行基准研究的科学家提供支持和信任。
莫里斯说:“对标论文是绝对必要的。“这是对社区的重要贡献。”
当开始单细胞研究时,研究人员必须做出的第一个选择是使用哪种技术分离细胞并分析其分子。对于单细胞RNA (scRNA)测序,一种常见的方法是首先将单个细胞分成孔或液滴,逆转录细胞的RNA以生成互补DNA (cDNA),然后使用分子条形码为每个孔的cDNA贴上不同的标签。接下来,每个细胞的cDNA被扩增以构建一个文库,然后cDNA链及其标记被测序。最后,条形码被用来确定RNA的哪个片段来源于哪个细胞。但科学家们有很多方法来完成这些任务。
测试技术
2017年,麻省理工学院和哈佛大学布罗德研究所的遗传学家和分子生物学家约书亚·莱文(Joshua Levin)与同事们一起比较了七种scrna测序方法。其中两种是低通量方法,适用于数百个细胞的分析,通常在捕获稀有RNA和细胞类型时提供高灵敏度。其中五种是可以处理数千个细胞的高通量方法。
研究人员将这些方法应用于三个样本1.Levin说,其中之一,人类外周血单个核细胞(pmcs)是一个很好的测试案例,因为科学家已经知道哪些细胞类型应该存在,以及它们的分子特征是什么样子。人类和小鼠细胞系的混合使研究人员能够检测到两个细胞落在一个孔中的情况。这将表现为人类和小鼠的基因共享相同的条形码,这是一个明显的错误指示。该团队还测试了四种适用于单核RNA测序的方法,这种方法可以很好地适用于难以分离单细胞的某些组织。在这些测试中,他们使用了小鼠脑组织,因为它是此类研究的常见目标。
理想的方法取决于科学家的数据集和研究问题。Smart-Seq2而且CEL-Seq2,这两种低通量方法的表现相似,在每个细胞中识别了最多的基因——尽管CEL-Seq2有时会将RNA序列分配到错误的细胞。莱文说,这两种方法的缺点是价格昂贵。在高通量方法中,10X Chromium系统效果最好,每个细胞提取的基因最多。该系统由位于加州普莱森顿的10X基因组公司开发,将样本分割成液滴。
Levin说,在进行基准研究时,最大的挑战之一是确保所有的方法都是公平的。为了控制实验之间的差异,研究小组在同一批次中准备了不同方法使用的样品。当他们进入测序步骤时,他们在机器的同一室中进行每种反应。他们还开发了一种计算管道,以尽可能相似的方式处理所有数据。莱文的团队自己执行所有的方法,但这意味着他们不一定是每种技术的专家。对于对他们来说是新的方法,研究人员向每种技术的开发者寻求帮助,以使其正确。

加州生物技术公司10X基因组公司研发基因测序技术。图源:Pavlo Gonchar/SOPA Images/Shutterstock
西班牙巴塞罗那国家基因组分析中心的生物技术专家霍尔格·海因领导的研究小组采取了不同的方法2.海恩的团队将单细胞和单核方法的测试外包给了专家,并将同一试管中的样本发送到世界各地的13个中心。Heyn说:“单细胞技术通常需要强大的专业知识和经验,才能正确地进行分析并获得尽可能好的结果。”
在他们的样本中,研究人员选择了人类pbmc和小鼠结肠组织的混合物。前者提供了明确定义的类型。结肠组织包括从干细胞向完全分化的结肠细胞发育过程中的细胞,提供了连续的细胞类型和大小。

单细胞多组学如何建立关系
为了进一步评估这些方法检测罕见细胞类型的能力,研究小组添加了定量的荧光标记的人类、小鼠和狗细胞系。这种特定细胞类型的“激增”,以及不同种群的混合,使科学家能够将他们的观察结果与他们所知道的样本中存在的细胞的确切数量进行核对。例如,在Heyn的研究中测试的一种方法遗漏了构成细胞鸡尾酒1%的犬类细胞;另一项统计显示,犬类细胞太多,占人口的9%。
Heyn说,与将细胞分裂成液滴的微流体方法相比,在微滴板上的孔中分离细胞的方法通常能得到高质量的结果,但吞吐量有限。CEL-Seq2和另一种方法Quartz-Seq2都擅长发现基因,尤其是CEL-Seq2甚至可以检测到弱表达的转录本。但Quartz-Seq2总体上表现最好,因为它在通过已知生物标志物的表达对细胞进行分组时得分也很好。
评估算法
一旦科学家决定了一种分析方法,他们必须选择一种方法来将原始数据转化为有意义的结果。同样,还有很多选择。一些算法获取rna测序数据,并按类型对细胞进行分组,或者检测反映一种细胞类型向另一种细胞类型发展的基因表达进程。其他算法评估DNA序列以确定染色质结构,分析转录因子的作用或评估用于细胞-细胞通信的分子。有些甚至结合不同实验的数据,修正不同时间或不同研究人员分析的批次之间的差异。
瑞士苏黎世大学(University of Zurich)的计算生物学家马克·罗宾逊(Mark Robinson)说,基准测试是根据分析技术执行任务的准确程度来评估的,但具体的指标取决于手头的任务。例如,科学家的基准测试技术旨在将相似的细胞类型聚类在一起,可能会检查“F1分数”,它包含了正确分类的数量,以及该数字与可能的正确分类总数的比较。相比之下,旨在量化差异基因表达准确性的科学家可能会选择统计力,即检测这种差异的概率的一种度量。基准测试还可以评估效率、计算需求、文档质量以及算法背后的代码是否开源等指标。

单细胞分析进入多组学时代
科学家可以使用两种输入数据集:来自细胞的真实数据,以及由诸如飞溅这是一个可以模拟scrna测序数据的软件包。Sandve说,模拟数据提供了一个很好的起点,因为它们提供了一种“健全检查”——研究人员准确地知道他们在管道中放入了什么,所以他们知道应该出现什么。
但是模拟数据永远不能代表真正的生物复杂性,因为研究人员无法模拟他们不完全理解的复杂性,Kim-Anh Lê Cao说,他是澳大利亚帕克维尔墨尔本大学的计算统计学家。真正的细胞具有真实的复杂性。“缺点是,”萨伊斯-罗德里格斯说,“我们可能对正在发生的事情没有一个明确的基本真相。”
理想的情况是,科学家们用黄金标准的数据集来测试分析方法,在这些数据集中,细胞类型和生物学特征得到了很好的表征,因此他们可以预测结果。该数据库深度集成了人类单细胞组学信息,称为迪斯科,包括来自各种组织类型、疾病和分析平台的已发表数据集。
根据Robinson在9月份发布在预印本库bioRxiv上的一项元分析,已经完成了60多项单细胞计算方法的基准研究3.因此,寻找理想方法的科学家有很多选择。Sandve建议,寻找中立的分析,因为新算法的开发者自然会强调他们方法的优点。然后,专家建议,科学家们应该寻找与自己的数据集相似的方法,并在确定最佳方法之前,用自己的数据测试一些方法。
最复杂、最先进的方法并不总是最好的。例如,麻省总医院和波士顿哈佛医学院的计算生物学家Luca Pinello与剑桥哈佛大学的表观遗传学家Jason Buenrostro合作,他们的同事比较了几种基于染色质可及性(与基因表达相关的测量)来区分细胞类型的方法4.当他们绘制计算运行时间与结果质量的关系时,他们惊讶地发现两者之间没有关系。皮内洛说:“有时候,做非常复杂的事情并不能提高你的表现。这意味着计算资源有限的科学家仍然可以找到能给出良好结果的算法。
有时答案不是选择一个单一的,最好的方法,而是使用几个。这就是Saez-Rodriguez和他的同事们得出的结论,他们对从scrna测序数据推断细胞-细胞通信途径的方法进行了基准测试,发现它们之间的一致性不高5.他说,如果多种分析途径给出了相同的结果,那么结果可能是正确的。他的团队开发了一个开源框架叫做藤本植物它允许用户在几个数据集上运行多个算法,允许公平和公正的比较,赛斯-罗德里格斯说。该系统还可以从所有包含的方法中提供一致的结果。
这需要一个村庄
尽管基准测试的价值显而易见,但科学家们表示,这类研究既没有得到特别充足的资金支持,也没有受到高度重视。布恩罗斯特拉哀叹道:“学术界并没有因为这类活动而得到回报。”
但这种情况正在开始改变。该研究所计算生物学科学项目经理伊凡娜•耶利克(Ivana Jelic)表示,位于加州雷德伍德城的陈•扎克伯格计划(Chan Zuckerberg Initiative)已经在单细胞生物学的基准测试和相关研究上投入了数千万美元。她补充说,获得认可的机会正在增加。
例如,布恩罗斯特罗对最近采用的一种纸质格式感到兴奋自然方法它寻求奖励基准研究。被称为登记报告,该格式邀请研究人员在收集数据之前提交他们的计划。如果投稿符合期刊的新颖性、范围和全面性的标准,它将被暂时提前接受。在数据收集之后,如果研究通过了质量检查,并以杂志认为合理的方式解释了研究结果,那么无论结果如何,它都将被发表。布恩罗斯特拉说:“这在这个领域非常非常有价值。

NatureTech中心
研究人员还可以联合起来进行基准测试。2019年,德国Neuherberg亥姆霍兹慕尼黑研究所和慕尼黑路德维希·马克西米利安大学的几十名研究人员聚集在德国度假胜地施里尔湖。他们的目标是测试各种方法来整合和规范化来自不同来源的数据,这对研究人员建立大型单细胞数据集至关重要。在团队中,该小组处理了来自120多万个细胞的RNA序列和染色质可达性的模拟和真实数据。在国内进一步努力之后,他们的研究于去年12月发表——距离收集数据已经过去了两年多6.在单细胞技术发展的世界里,这是一段很长的时间。随着工具的不断出现,任何单细胞基准测试纸的保质期只有几年。
马修·里奇(Matthew Ritchie)是澳大利亚帕克维尔沃尔特和伊丽莎·霍尔医学研究所(Walter and Eliza Hall Institute of Medical Research)的生物信息学家,他没有参与这项研究,他说:“随着技术的变化,协议的变化,一切都在改进,软件也在变化,所有这些都发生得很快。”“有必要让这成为一个持续的过程。”
但对于研究人员来说,通过添加一两个新方法来简单地更新基准研究通常是不可行的。罗宾逊在他对62项研究的元分析中说道3.他发现,每项研究都倾向于使用自己的代码,没有任何标准或系统化。他说:“几乎不可能使用这些代码并对其进行扩展和构建。”
为了解决这一缺点,Robinson和其他人正在构建类似于LIANA的伞形系统,允许用户使用各种金标准数据集来比较算法。一个系统叫做单细胞分析中的开放问题,提供了一个“活生生的基准平台”,该网站的联合创始人、马萨诸塞州萨默维尔制药公司Cellarity的机器学习科学家丹尼尔·伯克哈特(Daniel Burkhardt)说。用户可以访问排行榜,对不同数据集上的方法进行排名;目前已有数据去噪、批处理集成等十大类功能,包括十多个数据集和大约四十几种方法。合作者运行所有的分析,并提供代码供用户下载;方法开发人员可以提交工具,并为他们自己的方法对代码进行调整或改进。伯克哈德说,自2021年1月上线以来,该网站已有2.5万名独立访问者。
他说:“以集中式方式进行的基准测试要比在论文发表时进行的基准测试好得多。”其他系统的选择包括罗宾逊系统OmniBenchmark,OpenEBench来自ELIXIR工具平台。但这些产品尚未起飞;罗宾逊指出,在他的元分析中的62篇基准论文中,没有一篇研究使用了该系统。
研究人员预测,随着时间的推移,发展将放缓,生物信息学家将趋同于更标准化的方法,就像新技术经常发生的那样。
“这是一场疯狂的竞赛,”Lê曹说,“但你已经可以看到,有些人倾向于使用相同的工具。并不一定是因为它们是最好的,而是因为它们很容易使用。”该领域仍然需要时间来确定一个或多个制胜策略。目前,基准测试可以帮助科学家们在面对一系列令人眼花缭乱的选择时缓解困惑。
 单细胞多组学如何建立关系
单细胞多组学如何建立关系 单细胞分析进入多组学时代
单细胞分析进入多组学时代 海星事业:在单细胞中寻找RNA模式
海星事业:在单细胞中寻找RNA模式 NatureTech中心
NatureTech中心