
文摘
走出非洲的主要事件之后,人类与尼安德特人交配留下1 - 2%的尼安德特人DNA散落在今天在非洲以外的所有基因组小片段。这里我们调查可以了解人类人口的过程从这些片段的大小分布。我们观察到的差异片段长度横跨欧亚大陆有12%长片段在东亚人比西方欧亚混血。对比现存人口对于古代的样本显示,这些差异是由不同的速率衰减长度的重组以来,尼安德特人掺合料。在和谐中,我们观察到很强的相关性之间的平均片段长度和突变积累,类似预计通过改变繁殖的年龄估计从三人的研究。总之,我们的结果表明不同世代间隔横跨欧亚大陆,由10 - 20%,在过去的40000年。我们使用性别变异特征可以推断这些变化是由男性或女性生殖年龄的变化,或两者兼而有之。我们还发现,之前报道的突变谱的变化可能主要通过世代间隔的变化来解释。我们认为尼安德特人的片段长度差异提供了独到的见解,人类在最近的历史。
介绍
如果在所有来自非洲的一个尼安德特人的序列基因渗入的事件,然后在穴居人的差异片段长度分布世界各地将表明重组的速度差异时钟在人群中。假设固定数量的每一代起,这就意味着不同掺合料事件以来一代又一代的数量,因此代次的差异数量。虽然最近的研究指向一个基因流的事件1,一个额外的掺合料事件私人亚洲人也被提出2,3,4因为亚洲携带大量的尼安德特人的基因组序列相比,欧洲的基因组。然而,亚洲也将有更多的古老的基因组片段,如果一个基因流共同欧亚混血之后,稀释的尼安德特人的内容在欧洲,由于后续没有尼安德特人掺合料掺合料人口1,5。
一个独立的信息来源估计世代时间的差异是派生的等位基因积累的速率和频谱基因组在给定的时间6,7。谱系的研究表明,每年突变率稍微增加当世代时间减少,因为突变破裂在青春期前的生殖系代表一个高比例的年轻父母的新突变8。此外,每个突变类型的相对比例取决于繁殖父亲和母亲的年龄。这是利用估计的差异生成间隔的尼安德特人与人类之间的男性和女性6。
这里我们研究古老的片段长度分布在现存的非洲的基因组从西蒙斯基因组多样性项目(SGDP)9和六个高覆盖率古老的基因组。我们报告的证据一个尼安德特人掺合料事件由所有欧亚和美国共享个体,使我们能够利用古老的片段长度分布的测量每隔一代因为掺合料。估计代间隔的差异反映出突变的积累和整合模式显示显著差异在经历的一代时间间隔不同的欧亚地区自40000年前他们的分歧。
结果
尼安德特人的片段长度分布横跨欧亚大陆不同
平均的片段长度从SGDP非洲以外的人,使用的方法推断Skov et al。10横跨欧亚大陆和美国有所不同(无花果。1、方法、S1 Data1_archaicfragments.txt)。提出了一个明确的西梯度意味着片段长度最低的个体从中东(S_Jordanian-1,意味着= 65.69 kb, SE = 2.49 kb, sd = 72.09 kb,方法)和从中国最高的个体(S_Tujia-1,意味着= 88.70 kb, SE = 3.29 kb, sd = 110.62 kb,方法,补充图。5)。时定性模式是非常相似的)中间片段长度而不是意味着使用长度,b)限制片段最密切相关的出土的尼安德特人的基因组测序的尼安德特人最密切相关的人口introgressing尼安德特人11或c)只使用高信任度模型推断的碎片(补充图。2- c,S2)。当个人被分成五个主要地理区域、古老的片段长度平均分布明显不同(P值< 1 e−5排列测试方法)(图1.12倍。1 b放大,补充表2)。非常相似的和显著差异也在人类基因组多样性计划独立(HGDP)1从每个区域数据相比更同质的人群(撒丁岛人,拉祜族S5, Data3_HGDParchaicfragments.txt)。这五个地区也显示显著差异数量的古老的碎片和陈旧的序列推断每个(P值< 1的e-5、排列测试方法,无花果。1 c, d补充表2),镜像的意思是古老的片段长度分布模式。与之前的报道相一致1,4,我们发现东亚人1.32倍更古老的序列推断每个相比西方欧亚混血(P值< 1 e−5排列测试方法,无花果。1 d补充表2)。
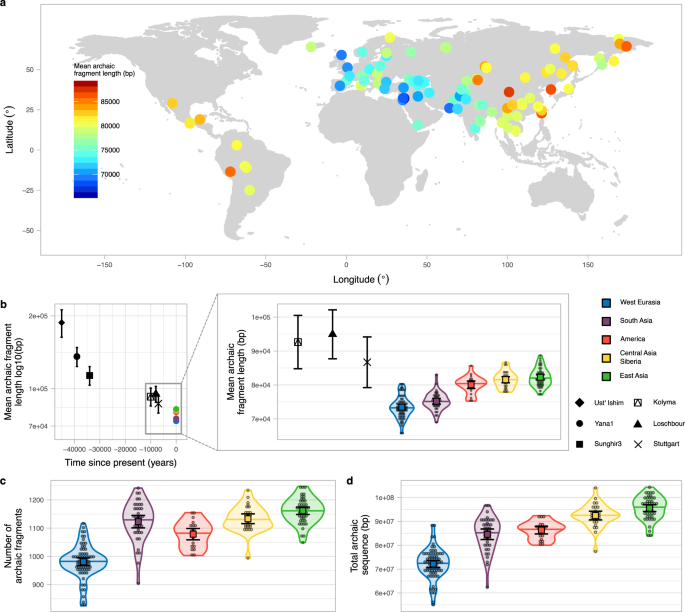
一个世界地图(方法)显示圈SGDP样本用于研究彩色根据意味着古老的片段长度。b意味着古老的片段长度的现存的地理区域和古代样本。科大的-Ishim、Yana1 Sunghir3科累马河,Loschbour和斯图加特的意思是古老的片段长度显示为黑色的点与特定的形状误差与相应的95%置信区间。汇总统计数据的样本大小的碎片,连同其他的统计数据,在补充表表示3。古老的片段长度的平均值在所有个人的五个主要地区显示为点(红、黄)。的放大显示的意思是每个地区古老的片段长度分布(颜色编码)作为小提琴阴谋。个别值显示为点。中值显示为一条水平线在每个小提琴阴谋。每个分布的均值及其95%可信区间显示为彩色广场与相应的误差。个人的样本大小为每个区域汇总数据导出,连同其他统计,在补充表表示2。科累马河,Loschbour和斯图加特的意思是片段的长度也显示比较。c,d古老的碎片的数量和陈旧的序列分布,分别,每地区(颜色编码)小提琴阴谋。个别值显示为点。中值显示为一条水平线在每个小提琴阴谋。每个分布的均值及其95%可信区间显示为彩色广场与相应的误差。个人的样本大小为每个区域汇总数据导出,连同其他统计,在补充表表示2。
我们下一个调查是否更大数量的古老的序列在东亚人通过不同的古老的片段解释由于第二个尼安德特人的外加剂。我们通过加入45东亚人的片段和比较他们的加入片段子样品45西方欧亚个人(补充图。7 a、b、方法、补充图。8)。总共有916369 kb的基因组是由古老的序列在东亚和866945 kb在欧亚大陆西部,有485255 kb(分别为53和56%)古老的序列重叠(无花果。2补充表6)。因此,作为一个群体,东亚地区只有6%的基因组位置与古老的基因渗入私人陈旧的证据和不存在多余的序列,预计将从私人脉冲(方法,S7,请参阅下面的模拟研究)。如果我们进一步删除片段与最近的亲和力排序丹尼索瓦人(S6),东亚人具有更多的特性12,总由古老的片段序列几乎相同(东亚853065 kb,欧亚大陆西部850028 kb,补充表8)。当我们限制与亲和力的片段,克罗地亚Vindija或阿尔泰尼安德特人(S6),东亚有更高比例的基因组7%(东亚646710 kb,西方欧亚混血604518 kb,补充表8)。我们把后面的这个区别,西方欧亚混血的短片段都让他们稍微难推断Skov et al。10方法也不太可能携带单核苷酸多态性(snp),直接把它们归入靠近克罗地亚Vindija尼安德特人。
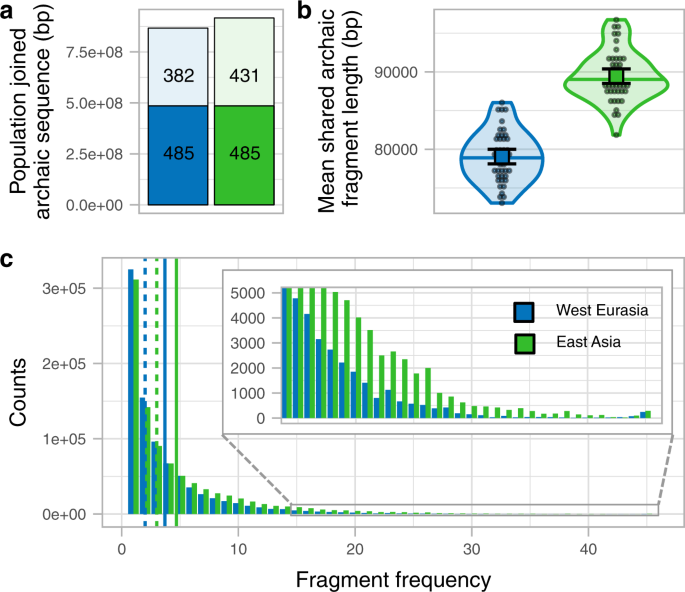
一个加入的顺序在地理区域(颜色编码)。酒吧在素色漆的部分显示了共享数量之间的区域。其余的列显示每个区域的序列私人。每个部分的数字表示相应的古老的序列Mb。个人的样本大小为每个区域汇总统计数据来源于,连同其他的统计数据,在补充表表示6。b个人共享的意思是古老的片段长度分布片段在地区/地区(颜色编码)小提琴阴谋。个别值显示为点。中值显示为一条水平线在每个小提琴阴谋。每个分布的均值及其95%可信区间显示为彩色广场与相应的误差。个人的样本大小为每个地区汇总统计数据来源于,连同其他统计,在补充表表示7。c的数量1 kb基因组窗口(y设在)中发现了一个古老的片段一定数量的个人(x为每个区域设在)。插入显示了高频箱。垂直线显示平均(纯线)和中值为每个地区(虚线)。个人的样本大小为每个地区汇总统计数据来源于,连同其他统计,在补充表表示7。
比较共享片段的长度,我们只考虑重叠碎片在东亚地区西部欧亚混血的基因组包含古老的序列,反之亦然(补充图。二维补充图。7 c、方法)。我们观察到共同的碎片在东亚人平均1.13倍的时间比在西方欧洲人群(P值< 1 e−5排列测试方法,无花果。2 b补充表7),也观察到当碎片都是上面使用。
基于这些观察结果,我们得出这样的结论:绝大多数甚至所有的尼安德特人的祖先在东亚和西方欧亚混血源于相同的尼安德特人掺合料事件,由组级别分析如图所示。这是由模拟表明额外的尼安德特人掺合料为东亚人变化意味着片段大小仅略(2.5%),导致过多的私人东亚的碎片没有观察到在我们的分析(S7)。总额32%的更大的古老的序列在东亚与西方相比欧亚个人平均主要是由于陈旧的片段出现在东亚人(图更高的频率。2摄氏度补充图。7 d补充图。8)。频率不太可能发生的转变通过自然选择作用更强烈反对陈旧的片段频率在欧亚大陆西部,清除以来的尼安德特人基因渗入之前预计是分裂的欧洲和亚洲的人口13,14。我们考虑我们的观察更符合欧洲人的混合人口基底欧亚很少或根本没有陈旧的内容稀释了尼安德特人的祖先曾建议使用古老的样品从外加剂的造型5。这样一个稀释的过程将会转变他们的频率分布我们观察(图。2摄氏度)。然而,它的影响将是微乎其微的尼安德特人的平均长度片段,因此能不能解释长度之间的差异我们观察西方欧亚大陆和东亚(无花果。2 b)。
复合景观的差异数量可能会影响到古老的片段长度分布。我们使用特定人群精细复合地图15推断出的物理长度片段转化为复合长度(方法、S4)。我们发现西方欧亚大陆和东亚之间的差异定量非常类似的片段以碱基对(补充无花果。3,S4)。这让我们以不同的速度复合时钟自常见的掺合料与尼安德特人是最古老的片段长度分布差异的可能原因。因此,我们建议改变一代间隔的最可能的原因是我们看到的片段差异。
古代回让我们看看古老的基因组片段长度。我们在六高覆盖率古代样本称为古老的碎片16,17,18,19,20.45000至7000年前之间约会(方法、S1补充表1补充图。1补充表3)。正如预期的那样,所有古老的样品比现存种群平均再古老的碎片和样本日期接近的尼安德特人基因渗入事件有较长的意思是古老的片段大小推断(图。1 b补充图。1补充表3也看到傅et al。16和Moorjani et al。21)。然而,Stuttgart-which欧亚人口是农民直接关系到西部区域的重叠意味着间隔最长的片段(无花果。1 b放大)。这表明东亚人群,例如,必须经历了类似的复合比西方的祖先欧亚混血,这里代表斯图加特样本。因此,西方欧亚混血和东亚之间的区别意味着对应100 - 370代(假设平均世代时间29年)在大约40000年分裂的欧洲和亚洲的人口1,22,23,24。
突变积累不同横跨欧亚大陆
新创突变的数量(认为)传送到一个孩子依赖于父母的性别和年龄8。因此,世代时间的变化在最近的人类进化历史,如上建议,应该留下一个突变累积总数的检测模式。为了验证这一点,我们估计衍生等位基因的数量积累在每个单独的常染色体分裂以来,非洲和非洲以外的人群(方法、S8 Data2_mutationspectrum.txt)。这是通过首先删除派生的等位基因在撒哈拉以南非洲地区外,不包括那些与西方探测欧亚血统9。此外,我们掩盖所有的基因组区域与古老的基因渗入的证据在任何个人在这项研究中,因为陈旧的变异不会发现在撒哈拉沙漠以南的基因组和他们会影响我们的结果,因为他们积累下不同的突变过程6。屏蔽这些地区也保证了这个分析是独立于古老的片段长度分析上面。这些过程后,我们只剩下~ 20%的可调用的基因组(S8)。
图3显示的速度积累衍生等位基因明显不同的组间(P值= 3 e−4、排列测试方法,补充表9)。欧亚大陆西部积累了1.09%比东亚衍生等位基因(P值= 1.3 e−3、排列测试方法)自从走出非洲的事件。然而,这种差异在衍生等位基因的积累只可能发生在西方欧亚大陆和东亚是分开的,这只是一个时间自走出非洲的一部分(S9,补充图。13)。如果我们假设> 60000年走出非洲和亚洲西Eurasia-East < 40000年的分裂1,22,23,24(S9)派生的等位基因的差异累积至少60000/40000×1.09% = 1.64%,West-Eurasia和东亚(方法)。使用pedigree-based估计之间的关系意味着每一代父母年龄和变异率8(方法),我们估计,这种差异对应于一个短2.68或3.39年世代间隔在欧亚大陆西部如果东亚意味着世代时间分别是28或32年(方法、S9)。这些下界推断不同的一代的间隔自走出非洲的区别和人口分裂时代最小化了。
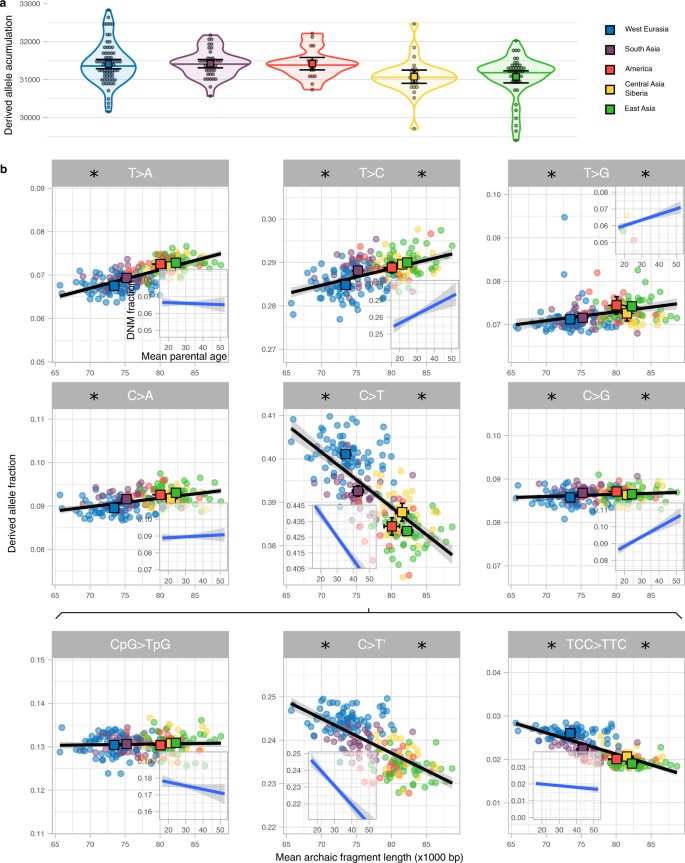
一个分布的衍生等位基因积累(y设在)地区(颜色编码)小提琴阴谋。个别值显示为点。中值显示为一条水平线在每个小提琴阴谋。每个分布的均值及其95%可信区间显示为彩色广场与相应的误差。个人的样本大小为每个地区汇总统计数据来源于,连同其他统计,在补充表表示9。b派生的等位基因之间的相关性(比例y设在)的意思是古老的片段长度(x设在)九突变类型。每个点代表一个单独的颜色根据他们属于该地区。对于每个区域,两轴及其95%可信区间是显示为彩色与相应的误差。个人的样本大小为每个地区汇总统计数据来源于,连同其他统计,在补充表表示2和补充表10。线性回归(黑线)显示相应的SE(阴影区域)。对于每一个突变,分数之间的线性回归和相应的SE认为,父母年龄和平均每渊源者解码数据(方法、S10)显示为一个插入。注意的总时间y设在是相同的所有面板和插入但特别集中在中值在每个面板和插入。星号的左右每个突变类型表明,线性回归显著不同的斜率从0 SGDP和解码数据,分别为(补充表11)。
父母的年龄概念,因此生成时间,也影响类型的单核苷酸突变发生的频率8。因此,转变一代时间预计新突变的光谱变化6,7和部分解释人群中描述的突变谱的差异25,26,27。我们计算的相对频率六种不同类型的单核苷酸突变取决于他们的祖先和衍生等位基因(方法、S8补充图。12补充表10)和相关的尼安德特人的片段长度平均为每个单独的(无花果。3 b)。我们观察到显著的关联平均陈旧的片段长度为所有六个类型(补充表11)。我们进一步细分C > T突变为以下三种类型:CpG > TpG,呈现出明显的突变过程28,> TTC移行细胞癌,这是多余的在欧洲基因组,研究特定人群突变签名25,26剩下的,表示C > T”(无花果。3 b补充表11)。我们发现CpG > TpG的频率转换至少取决于片段长度。
调查是否这些相关性可能是由于地理区域之间世代时间的差异,我们认为可利用的比例进行突变类型的函数意味着父母的年龄在解码三个数据集29日(无花果。3 b插入、方法、S10补充表11)。比较SGDP数据的相关性和解码数据我们看到一个强大的通信对于大多数突变类型:在所有类型的相关性与数据集意义重大,影响整合的方向(图3 b)。此外,我们与山坡上估计在两个数据集,以定量研究为每个突变类型相似的线性模型。我们找到一个关系接近一对一通信(斜率= 1.058,P值= 1.77 e−3,补充图。14)表明类似的突变谱模式变化多态性数据比单一代突变数据与世代时间的估计。解码的数据集有一个轻微的偏向渊源者有年长父亲比母亲(= 2.77年,sd = 4.25,补充图。15),这可能影响响应突变类型的分数取决于意味着父母年龄。但是,没有重大变化相关系数是观察当只有渊源者类似父母的年龄分析(S10,补充图。16)。
由于没有先验理由期望一个古老的片段长度和衍生等位基因之间的关系积累,我们认为它可能相同的潜在因素影响。一般认为这些相关性与预期研究的通信支持我们的假设,这因果元素是一个世代间隔的变化。更具体地说,与父母年龄匹配的减少相关的移行细胞癌> TTC突变表明该突变签名时将增加意味着父母年龄下降。因此相当大的减少意味着在西方欧亚混血,世代时间,建议在这项研究中,提供了另一种解释多余的移行细胞癌> TTC突变与世界其它地区相比,该地区26,30.。
增加意味着世代间隔可以是由于父亲或母亲的年龄的增加,或者两者兼有。人类学研究表明,男性一般都比女性在生殖,但年龄差距是狩猎采集者与久坐的人群的两倍31日。了解性别改变一代时间间隔我们首先比较常染色体之间的派生突变的积累,这花相同数量的进化时间在两种性别中,X染色体,2/3的时间花在女性1/3在男性(S11)。因此,增加的相对男女世代间隔预计将增加X染色体常染色体(X-to-A)突变积累比例32,尽管生殖等其他因素方差和人口规模的变化也可以影响比率。图4显示了X-to-A衍生等位基因比积累每个碱基对(方法)的函数意味着古老的片段长度,作为女性平均一代时间代理,SGDP数据。我们观察到X-to-A地区之间的比率明显不同(P值= 4.4 e−4排列测试方法)。东亚人X-to-A更高比率相比美国和中亚和西伯利亚与尼安德特人的片段大小相似,和高于西方欧亚混血,较小的尼安德特人的片段大小。这个结果是符合东亚人平均一代时间高于西方欧洲人群主要是由于父亲的年龄在母亲的年龄增加繁殖相比美国和中亚和西伯利亚两性的生殖年龄推断增加了更多类似的。但是,我们承认这个测试仍然是有限的数据量,因此结论是初步的。

另一个性别突变签名C > G基因的突变区域集群认为在老母亲8,33。这个签名可以探讨产妇年龄组间进行比较6。我们估计的比例提取C > G等位基因在这些基因组区域其他衍生等位基因类型,对比相同比率的基因组,为每个单独的(方法、S11)。当样品被分组在5个主要地区,认为集群的C > G比例显著不同(P值= 3.1 e−3、排列测试方法),尼安德特人的片段长度的增加而增加(图4 b)。值得注意的是,美国有一个更高的比率比中亚和西伯利亚的尼安德特人的片段长度相似,显示一个相对较大的老母亲整体平均世代时间的影响在他们的历史。这是符合X染色体分析,再代次在美国更受年长的母亲相比,年长的父亲在东亚的中间增加父母的年龄在中亚和西伯利亚。
最后,Y染色体预计也将积累更多衍生等位基因在群体与年轻的父亲,同样常染色体,大约每年0.4 - -0.5%两个种群之间世代时间的差异。我们观察到1.19%的点估计西方欧亚大陆和东亚之间的较大的积累(S11,补充图。17补充表13),但这不是重要的有限的数据可用的Y染色体(P值= 0.66,排列测试方法)。
讨论
我们已经表明,尼安德特人在现代人类基因组片段的长度可以用来获得基本的人口统计信息参数,平均世代间隔。我们估计大跨欧亚和美国组差异表明稳定分歧成千上万年。我们的方法取决于假设陈旧的碎片追溯到一个尼安德特人掺合料事件共享的所有非洲以外的人群,我们提供进一步的证据。符合这些结果,推导出突变的数量积累在地理区域研究按照预期的世代时间估计的差异片段长度。重组和突变时钟之间的协议签名反对的混杂因素。例如,一个潜在的偏见会预期如果非洲群外,用于寻找古老的碎片在另个人,经历过一些古代基因流从西方欧亚大陆,我们无法检测到。这样的场景将缩短和删除陈旧的碎片在西欧亚混血,解释观察到的梯度。然而,这也将减少衍生等位基因的数量在欧亚大陆西部相比,东亚,我们报告的相反。
不同世代间隔的大小和持续时间,我们估计可以解释观察到的人类种群的突变谱的变化没有根本改变分子状态突变的结果在每一代的因素,即调整器和antimutators。这方面的一个例子的太极拳> TTC突变的频率增加西方欧亚混血。世代时间的差异,推断出从古老的片段长度,超过一半的解释总变异个体(调整R2= 55.42%)。
对先前的调查结果有直接影响的人类人口参数,通常假定世代间隔是共享和常数不同的人群。因此,未来的研究应该考虑变化生成时间。我们没有一个解释大型世代间隔差异的根本原因,但是,这是合理的环境、技术和文化背景的人群影响父母的年龄他们的后代。越来越多的古代和现代的基因组测序,我们预计,这里我们提出的方法可以用来获得一张精细的世代间隔的变化在过去40000年的时间里,可以直接关系到人口密度的变化、气候和文化。
方法
均值和置信区间的计算
均值及其置信区间(CI)的统计计算使用的均值和标准差100000引导观察统计量的抽样分布。计算它们的代码在GitHub页面上提供34。
排列的统计显著性评价测试
统计学意义的统计比较不同群体评估通过对比观察到零分布与非参数统计。零分布排列所产生的原始数据和计算统计的100000倍每一排列。
P值计算的分数排列,产生一个值作为极端或比什么是观察到的数据更极端。如果没有这样的事件中观察到所有的排列,我们认为分数是< 1/100,000 = 1 e−5。显著性水平(\α(\ \)在所有的测试被认为是0.05)。
测试如果有两组之间的差异的统计数据(例如,平均陈旧的片段长度),减去每组的方法。在这种情况下,因为这个测试是一个双尾假设检验,我们用P由两个值。当我们测试多个人群的差异,计算F统计。
计算统计显著性的代码是GitHub页面上提供的34。
识别陈旧的碎片在非洲以外的个人和古老的样本
我们称之为古代SGDP个人的碎片9从欧亚大陆和美国地区,6 HGDP古代现代人类和4人群1所述Skov et al。6,10——一步一步的教程也可以https://github.com/LauritsSkov/Introgression-detection。
简而言之,方法首先删除一组变量(snp)出现在一群没有假定陈旧的外加剂(撒哈拉以南非洲人口)的样品我们要检测的片段(非洲)。然后,考虑到window-specific突变率和callability方法将重叠窗口分为古代祖先和non-archaic祖先根据衍生等位基因密度。
更详细的信息的细节在调用陈旧的碎片在每个数据集可以在S1和S5。
世界地图的情节
使用R函数获得的背景地图边界,ggplot2库的一部分,从地图中检索地图映射函数库。后者进口1:50 m范围内公开的世界地图栅格自然地球工程。
古老的片段物理长度转换使用特定人群遗传距离复合地图
均值的片段长度5中不同地理区域的SGDP数据(正文)。为了测试特定人群复合地图在古老的片段大小的差异来解释,我们观察到,我们把片段的长度从物理单位(碱基对,bp)遗传单位使用特定人群(厘摩,厘米)复合地图推断phyro151000人基因组计划(1 kgp)和比较结果分布地区之一。
我们下载的数量的复合地图1 kgp35来自:https://github.com/popgenmethods/pyrho human-recombination-maps。补充表4显示了人口的子集分析在这项研究中,在1 kgp表示,因此,复合地图,我们可以使用。
我们注意到,对于不同人群,染色体有不同的基因在下载地图长度;一般来说,东亚人群倾向于推断长染色体。如果我们直接应用这些地图,它将导致东亚的片段被更长时间比其他人群基因长度比物理长度。因为没有理由相信,染色体遗传图谱的长度是不同的在人群中,我们按比例缩小的复合率,这样所有人口都有相同的总长度为每个染色体在遗传距离计算片段的大小。
对于每个染色体(c),我们首先规模重组率(r每个人口()p)的长度(l)的染色体的人口与最短的大小(问)
然后,更新率(r '),我们计算平均重组率(\(酒吧\ {r} \)一个古老的片段)(f)通过交叉复合块(k)和计算均值,权重每个块的重组率的复合块重叠与古老的片段之间的比例和总陈旧的片段长度(w)
最后,我们将每个片段的长度(年代)在物理单位(bp)遗传单位(厘米)使用意味着重组率(\(酒吧\ {r} \)每个片段)
补充图。3显示的意思是古老的片段长度分布在生理和遗传距离。比实际长度分布、基因长度分布总体少分散在区域和地区更大的区别。平均而言,东亚人比西方欧亚大陆有1.16倍的时间片段基因长度单位,这是一个更大的差异比相比,如果物理长度是1.10倍。然而,在这两种情况下,地区之间的差异具有统计学意义(基因长度P值< 1 e−5,实际长度P值= 4 e−5、排列测试方法)。这是由于良好的片段长度的两个指标之间的相关性为所有个人评估(补充图。4)。
我们得出结论,种群之间的遗传差异地图不能占片段长度描述的地区之间的差异。
西方欧亚大陆和东亚片段比较陈旧的片段基因组覆盖率
在主要的文本,我们比较碎片在西方欧洲人群和东亚人。个人用来恢复古老的碎片越多,可以找到更多的未被发现的碎片6。因此,个人的数量在每个地区的不平衡在SGDP数据(71年西方欧亚混血和45东亚人)可能会影响任何两个地区之间的比较。因此,我们downsample用于西方欧亚混血的个体数量45随机选择的个体比较公平。
首先,我们为每个地区加入所有重叠的片段,在此“加入地区碎片”(补充图。7)。要做到这一点,我们使用bedtools软件36(版本2.30.0)使用下面的命令:
bedtools我ind1_regx合并。床ind2_regx。bed … indN_regx.bed > joined_regx.bed在哪里x表示西方欧亚大陆或东亚地区N表示相应地区的人的数量。
然后,我们多少古老的序列相比这两个地区分享(补充图。7)。之间的相交,我们称之为两组加入的碎片。我们称它为“共享加入区域序列”。我们使用以下命令:
bedtools相交——joined_regx。bed -b joined_regy.bed > shared_joined.bed在哪里x表示西方欧亚大陆或东亚和y表示不同于其他地区x。
由此可见,其余的碎片没有包含在这个集合是“私人加入地区序列”。
序列的数量的共享,私人和总加入地区片段提供了补充表6。
对于每个人,我们分类的片段作为共享取决于是否有一个重叠的片段另加入地区片段(补充图。7)。我们的名字这些片段是“共享单个片段”。让他们,我们运行以下命令:
bedtools - u - indn_regx相交。bed -b joined_regy.bed > shared_indn_regx.bed
由此可见,其余的碎片没有包含在这个集合是“个人片段”。
摘要统计信息共享和个人片段提供了补充表7。
最后,我们计算个体的数量有一个重叠的片段基因组中某1 kb窗口。通过这种方式,我们计算“古老的频率”。,我们第一次加入区域中的每个片段片段划分为1 kb段(joined_regx_1kb.bed)。然后,我们统计数量的每个1 kb的重叠的片段段使用下面的命令:
bedtools相交- c——joined_regx_1kb。床- b ind1_regx。床ind2_regx。bed … indN_regx.bed > freq_regx.bed
补充图。8显示了加入地区摘要片段,共享加入区域序列和每个地区的古老的频率。
模拟
我们模拟使用msprime37整个基因组的两个人口场景:场景用一个尼安德特人的脉搏东亚和西方欧亚混血的共同祖先(一个脉冲)和另一个额外的和私人脉冲东亚人(两个脉冲,S7)。参数的场景补充图所示。9下面的列表和一些表示:
−−突变率= 1.25 e 8
−复合地图=人类基因组单体型图复合地图38下载http://bochet.gcc.biostat.washington.edu/beagle/genetic_maps/
−世代时间(年)= 29
为什么我们选择这些参数中解释S7。
对于每一个场景,我们执行十整个基因组的复制为了获得估计的方差统计分析。
在每个仿真,我们非洲集团的样本500人作为外围集团和50每个西方欧亚大陆和东亚。变异基因的位置然后转换为离散空间通过删除给定的浮动有多个变量的值为一个整数值和位置都被删除了。模拟类似的基因组callability概要文件在模拟数据,我们无视变异,落入不可赎回S1中使用的文件的位置。然后我们称为古老的碎片后,类似的方法来描述的一个部分和S1的方法。
这些模拟的详细的分析S7。
衍生等位基因叫外地区古老的基因渗入和获得的证据在SGDP走出非洲之后样品
我们检索所有多态位点的基因型为每个单独的5个主要地区和非洲样品使用cpo脚本的Ctools软件(1500年版)9对染色体22页。在参数文件中,我们指定的最小质量1(推荐Mallick et al。9)和等位基因与黑猩猩分化参考基因组(PanTro2) SGDP提供的数据。
接下来,我们蒙面重复区域和区域的基因组中有一些古老的基因渗入的证据。这是因为重复区域可能富含测序错误也因为尼安德特人比现代人类有不同的突变概要文件6。此外,通过消除这些区域,我们将基于基因突变分析的地区,我们还没有探索古老的片段长度研究的一部分。因此,测试是相互独立的。进一步的细节在这是如何进行的这项研究在S8解释道。
其他过滤器在SNP层面实施每个多态性等只保留biallelic SNP或派生的等位基因位点不隔离在非洲的事。进一步的细节在S8过滤器应用了。
派生的等位基因的纯合子轨迹算作2突变和杂合的网站算是1对于一个给定的个人。派生的等位基因的分布积累每地区图所示。3积累和平均等位基因数每个地区提供了补充表9。
我们分类在不同的突变位点类型取决于衍生等位基因核苷酸,祖先的等位基因核苷酸及其5′和3′核苷酸上下文,S8进一步解释。Data2_mutationspectrum。txt提供由此产生的重要的每个人在每个染色体突变类型。
估计不同的父母一代的时间在欧亚大陆西部和东亚
将衍生等位基因的过量积累在欧亚大陆西部相比,东亚地区在走出非洲之后,我们认为非洲《出埃及记》发生在60000年前,西方欧亚大陆和东亚种群之间的分裂发生在40000年前。这些保守的日期的原因分析是假定在S9详细。因为衍生等位基因的数量成正比走出非洲的事件,但只能积累过剩欧亚人群的分裂后,衍生等位基因之间的每分钱过剩积累(1.09%)必须根据这些时间扩大
在琼森等。8的泊松回归推导出突变的数量在每一代传播从三个数据为每个父母血统繁殖取决于他们的年龄
在下标\ \ (f)和\ \(米)分别表示父亲和母亲,{\ \(\帽子μ}\)的估计意味着每一代突变速率(\ (g \)),\ \ ()是指父母年龄。因此,假设是相同的父母年龄为祖细胞(f \({} _{} ={一}_ {m} = \)我们每一代总变异率计算的情商。3)和年度(\ (y \)情商的)速度。(4)。
然后,比较突变率每年在两个不同的种群(\ \ (x)和\ (z \))具有不同的意思是父母的年龄,我们得到
派生的等位基因的数量积累在基因组在一段时间(\ (d \)每年)取决于突变率和时间跨度(\ \ (T))
然而,的比例\ (d \)这两个种群之间只会依靠他们的突变率,因为\ \ (T)一直都相同。
因此,我们可以估计\ ({}_ {x} \)如果z \ ({} _ {} \)和(\ d {} _ {x} / {d} _ {z} \)是已知的
在这项研究中,我们发现的比例意味着衍生等位基因积累在欧亚大陆西部(\ ({}\))和东亚(\ ({EA} \),(\ d{} _{{我们}}/ {d} _ {{EA}} \))是1.0164 (1.64%)。与公式(8),我们检查合理\ ({}_ {{EA}} \)值28到32年,发现的值\({}_{{我们}}\)介于25.32和28.59之间。因此,我们估计代次东亚人一直在2.68 - -3.39年的时间比西方欧亚混血自分裂的两个群体。
突变谱相关性意味着父母的年龄
我们认为比较的突变模式取决于父母年龄在三人的研究8,29日(解码数据集)与现存种群的突变谱的差异意味着古老的片段长度的代理的意思是世代时间(SGDP数据集)。
类似琼森等。8,我们研究这些相关性如下线性模型的一般公式
在哪里\α(\ \)是拦截和β\ (\ \)线性回归估计的斜率。
SGDP数据集,我们分类衍生等位基因中发现每个人的常染色体6突变类型根据祖先和派生的等位基因在方法部分和S8解释道。C > T突变也分为三个亚型:> TTC移行细胞癌,CpG > TpG,其余(C > T”)。总的来说,我们把所有的突变分为九个类型。为了获得分数每个突变类型的个体,我们每一个突变类型的数量除以总数量的派生的等位基因。C > T突变是复制自我们细分为三个类别(> TTC移行细胞癌,CpG > TpG和C > T”)。因此,衍生等位基因总数不会考虑这三种类型。我们相关衍生等位基因的每种类型的分数意味着古老的片段长度的代理意味着生成时间(无花果。3 b)。我们获得了这种相关性的线性模型为每个突变类型使用以下函数R(补充表11)。
lm (mutation_fraction ~ mean_fragment_length)
解码的数据集,我们认为下载的集合称为Halldorsson et al。29日和额外的渊源者信息的出版物中提供补充数据(n认为= 2976的三人小组,200435)。我们加入这两个以计算平均为每个每个渊源者认为父母年龄。Indels被过滤掉。方法以类似的测试之后琼森等。8我们总变异数的九种不同类型的所有相同的渊源者意味着父母年龄。然后我们计算每个突变类型的一部分。换句话说,对于每个突变类型,父母年龄有一个突变分数值。那些得到的数据点从小于2渊源者被丢弃的聚合信息。我们获得的线性模型为每个突变类型使用以下函数R(补充表11)。
lm (mutation_fraction ~ mean_parental_age,重量= n_probands)
山坡上的两个数据集之间的相关性补充图所示。14。
性别突变模式
X-to-A比率是通过第一次计算派生常染色体的等位基因的数量和X染色体的女性SGDP数据(补充表12),在方法部分和所述S8(包括在Data2_mutationspectrum.txt)。然后,比率计算
在哪里\ (d \)表示衍生等位基因的数量,\ (L \)可调用的碱基对的数量\ \ (X)或(X染色体)\ \ ()(常染色体)。
计算C > G比值DNM-clustered地区(cDNM)和其余的基因组(non-cDNM),我们计算衍生等位基因的数量是C > G和非> G的基因组在windows 1 Mb。我们加入这个信息与基因组的注释1 mb-window cDNM或non-cDNM提供琼森等。8。然后,为每个单独的计算如下
在哪里\ (d \)表示数量的衍生等位基因C > G或非> G。因此,p两个量之间的比率。然后,比较之间的比率cDNM non-cDNM地区我们计算的意思\ (p \)(\(酒吧\ p {} \))在所有地区和计算比例如下:
如果\ (r \)= 1,这表明C > G浓缩cDNM地区相似比其余的基因组。如果\ (r \)> 1,然后有一个多余的如果\ (r \)< 1,然后有损耗。
数据可用性
SGDP的古老的碎片和它们的基本统计样本和古代Data1_archaicfragments.txt样品提供了源数据文件。96的数量每单个染色体突变类型Data2_mutationspectrum.txt源数据中提供的文件。陈旧的碎片和它们的基本统计数据HGDP Data3_HGDParchaichfragments样品提供了源数据文件。txt (S12)。所有三个源数据文件和本文一起提供。在这项研究中使用的所有数据是公开的。我们使用变体调用从个人全基因组测序提供的西蒙斯基因组多样性项目9(https://reichdata.hms.harvard.edu/pub/datasets/sgdp/),1000人基因工程35(https://www.internationalgenome.org/人类基因组多样性)和项目1(ftp://ngs.sanger.ac.uk/production/hgdp/)。我们使用了变体古代出土的尼安德特人的电话11,阿尔泰尼安德特人39和丹尼索瓦40(https://www.eva.mpg.de/genetics/genome-projects/)。最后,从基因组的测序读下面的古代现代人类存入ENA或SRA存储库进行分析:科大-Ishim16(PRJEB6622),Yana117(PRJEB29700和PRJEB26336),Sunghir318(PRJEB22592),Anzick119(SRX381032),科累马河17(PRJEB29700和PRJEB26336),Loschbour20.(PRJEB6272)和斯图加特20.(PRJEB6272)。
代码的可用性
生产数据和脚本编码表,进行统计分析和情节这手稿可以在Github(数据https://github.com/MoiColl/TheGenerationTimeProject)34。这个存储库中提供的脚本是MIT许可下的。
引用
Bergstrom, a . et al .洞察人类遗传变异和人口历史从929年不同的基因组。科学367年eaay5012 (2020)。
Villanea f . a & Schraiber j·g .多个集尼安德特人与现代人之间的杂交。Nat,生态。另一个星球。339-44 (2019)。
Vernot, b . et al .挖掘穴居人,丹尼索瓦人的基因组DNA美拉尼西亚人。科学352年,235 - 239 (2016)。
墙,j . d . et al .更高水平的尼安德特人的祖先在东亚洲人比欧洲人。遗传学194年,199 - 209 (2013)。
拉扎里迪斯,等。基因见解农业在古代近东的起源。自然536年,419 - 424 (2016)。
鉴于此,l . et al。尼安德特人的基因渗入的本质揭示了27566年冰岛基因组。自然582年,78 - 83 (2020)。
卡尔森,J。,德维特,w·S。&Harris, K. Inferring evolutionary dynamics of mutation rates through the lens of mutation spectrum variation.咕咕叫。当今。麝猫。Dev。62年50-57 (2020)。
琼森,h . et al .父母的影响对人类生殖系新创突变三人小组从1548年冰岛。自然549年,519 - 522 (2017)。
Mallick, et al。西蒙斯基因组多样性项目:300基因组从142年广泛的人群。自然538年,201 - 206 (2016)。
鉴于此,l . et al .检测使用一个unadmixed群古老的基因渗入。公共科学图书馆麝猫。14e1007641 (2018)。
Prufer, k等。高覆盖率穴居人基因组在克罗地亚Vindija洞穴。科学358年,655 - 658 (2017)。
布朗宁,s R。,Browning, B. L., Zhou, Y., Tucci, S. & Akey, J. M. Analysis of human sequence data reveals two pulses of archaic Denisovan admixture.细胞173年53 - 61。e9 (2018)。
哈里斯,k &尼尔森,r基因的尼安德特人基因渗入的成本。遗传学203年,881 - 891 (2016)。
切赫,M。,Pääbo, S., Kelso, J. & Vernot, B. Limits of long-term selection against Neandertal introgression.Proc。《科学。美国116年,1639 - 1644 (2019)。
斯宾塞,j . p . &歌,y s推理和分析特定人群精细复合地图在26个不同的人群。科学。睡觉。5eaaw9206 (2019)。
傅,问:et al。基因组序列的45000岁高龄的现代人类从西伯利亚西部。自然514年,445 - 449 (2014)。
Sikora m . et al .更新世以来人口西伯利亚东北部的历史。自然570年,182 - 188 (2019)。
Sikora, m . et al .古老的基因组显示早期旧石器时代晚期采摘者的社会和生殖行为。科学358年,659 - 662 (2017)。
拉斯穆森,m . et al。晚更新世人类的基因组从克洛维斯蒙大拿州西部的墓地。自然506年,225 - 229 (2014)。
拉扎里迪斯,et al。古代人类基因组显示三个今天的欧洲人的祖先种群。自然513年,409 - 413 (2014)。
Moorjani, p . et al .分子钟帮助估计年龄古老的基因组。Proc。《科学。美国113年,5459 - 5460 (2016)。
Schiffels, s &杜宾,r .推断人口规模和分离的历史从多个基因组序列。Nat,麝猫。46,919 - 925 (2014)。
傅,问:et al。DNA分析的早期现代人类从天元洞穴,中国。Proc。《科学。美国110年,2223 - 2227 (2013)。
Seguin-Orlando, a . et al . Paleogenomics。基因组结构在欧洲可以追溯到至少36200年。科学346年,1113 - 1118 (2014)。
哈里斯,k最近的证据,特定人群进化的人类基因突变率。Proc。《科学。美国112年,3439 - 3444 (2015)。
哈里斯,k &普里查德,j·k·快速进化的人类基因突变谱。Elife6e24284 (2017)。
马西森,i &帝国,d .人群中罕见的变异谱的差异。公共科学图书馆麝猫。13e1006581 (2017)。
Moorjani, P。,Amorim, C. E. G., Arndt, P. F. & Przeworski, M. Variation in the molecular clock of primates.Proc。《科学。美国113年,10607 - 10612 (2016)。
Halldorsson, b . v . et al .描述复合诱变的影响通过sequence-level遗传图谱。科学363年eaau1043 (2019)。
德维特,w·S。,Harris, K. D., Ragsdale, A. P. & Harris, K. Nonparametric coalescent inference of mutation spectrum history and demography.Proc。《科学。美国118年e2013798118 (2021)。
芬纳,j . n .跨文化人类世代间隔的估计人口用于基因差异研究。点。期刊。Anthropol。128年,415 - 423 (2005)。
amst, g . &鞍,g .生活史的影响中性多样性水平的常染色体和性染色体。遗传学215年,1133 - 1142 (2020)。
Goldmann, j . m . et al . Parent-of-origin-specific签名新创的突变。Nat,麝猫。48,935 - 939 (2016)。
科尔Macia,世代时间的项目。GitHubhttps://doi.org/10.5281/zenodo.5119901(2021)。
1000人基因组计划财团。一个全球参考人类遗传变异。自然526年,68 - 74 (2015)。
昆兰,a。r . &大厅,i m . BEDTools:一套灵活的工具来比较基因组的特性。生物信息学26,841 - 842 (2010)。
凯莱赫,J。,Etheridge, A. M. & McVean, G. Efficient coalescent simulation and genealogical analysis for large sample sizes.公共科学图书馆第一版。医学杂志。12e1004842 (2016)。
国际人类基因组单体型图财团。等。第二代人类单体型超过310万个snp的地图。自然449年,851 - 861 (2007)。
Prufer, k . et al。完整的阿尔泰山脉的尼安德特人的基因组序列。自然505年43-49 (2014)。
迈耶,m . et al .高覆盖率从一个古老的丹尼索瓦人个人基因组序列。科学338年,222 - 226 (2012)。
确认
感谢Felix Riede建议人类学的解释和马修·赫里斯建议对比衍生等位基因积累X和常染色体之间。我们感谢Juraj伯格曼和Marjolaine Rouselle所有讨论的后果尼安德特人稀释西方欧亚人群的场景。我们感谢Priya Moorjani评论和研究和提供反馈给有见地的建议。这项研究是由诺和诺德基金会的拨款NNF18OC0031004和6108 - 00385年M.H.S.研究委员会的独立研究
作者信息
作者和联系
贡献
M.C.M.,L.S. and M.H.S. designed the study. M.C.M. and L.S. created the methods to assess the data and, with M.H.S. analysed the results with input from B.M.P. M.C.M., L.S. and M.H.S. wrote the manuscript with comments from B.M.P.
相应的作者
道德声明
相互竞争的利益
作者宣称没有利益冲突。
额外的信息
同行审查的信息自然通讯由于席琳好,大卫·黑格和其他匿名审稿人,他们的贡献的同行评审工作。同行审查报告。
出版商的注意施普林格自然保持中立在发表关于司法主权地图和所属机构。
权利和权限
开放获取本文是基于知识共享署名4.0国际许可,允许使用、共享、适应、分布和繁殖在任何媒介或格式,只要你给予适当的信贷原始作者(年代)和来源,提供一个链接到Creative Commons许可,并指出如果变化。本文中的图片或其他第三方材料都包含在本文的创作共用许可,除非另有说明在一个信用额度的材料。如果材料不包括在本文的创作共用许可证和用途是不允许按法定规定或超过允许的使用,您将需要获得直接从版权所有者的许可。查看本许可证的副本,访问http://creativecommons.org/licenses/by/4.0/。
关于这篇文章

引用这篇文章
科尔Macia, M。鉴于此,L。,Peter, B.M.et al。在人类不同历史生成间隔推断从尼安德特人的片段长度和突变签名。Nat Commun125317 (2021)。https://doi.org/10.1038/s41467 - 021 - 25524 - 4
收到了:
接受:
发表:
DOI:https://doi.org/10.1038/s41467 - 021 - 25524 - 4
本文引用的
爸爸比妈妈大自人类的黎明,研究表明
自然(2023)
