
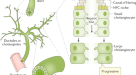





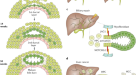


摘要
胆管细胞过度扩张(导管反应)促进肝脏疾病进展,但其潜在机制尚不清楚。在这里,我们发现胆道NF-κ b诱导激酶(NIK)是导管反应的关键调节因子。已知NIK可激活非规范IKKα/NF-κB2通路并调节淋巴组织发育。我们发现胆管结扎(BDL)、5-二氧羰基-1,4-二氢碰撞碱(DDC)或α-萘基异硫氰酸酯(ANIT)诱导的胆汁淤滞小鼠胆管细胞NIK上调。DDC、ANIT或BDL在小鼠中诱导小管反应、肝损伤、炎症和纤维化。胆管细胞特异性缺失尼克,但不是IKKα使这些病理改变变得迟钝。NIK抑制剂治疗同样可以改善ddc诱导的导管反应、肝损伤和纤维化。胆道NIK直接增加胆管细胞增殖,抑制胆管细胞死亡,并促进胆管细胞分泌胆管因子。胆管因子刺激肝巨噬细胞和肝星状细胞,增加肝脏炎症和纤维化。这些结果揭示了促进肝脏疾病进展的NIK/导管反应轴和NIK/胆管因子轴。
介绍
胆管为胆汁从肝脏流向胆囊提供通道,在这一过程中,胆管细胞——胆管上皮细胞——通过吸收和/或分泌各种分子来改变胆汁的成分。胆道损伤触发胆管细胞增殖以补偿胆管损失并维持胆道稳态。双能性胆管细胞样细胞可分化为胆管细胞或肝细胞,支持肝脏再生1.然而,过度的胆管细胞扩张导致致病性导管反应,这是胆汁淤积的标志,有助于肝脏疾病的进展2,3..胆管反应预示预后不良,增加胆管癌的风险3.,4,5,6.胆管结扎(BDL)诱导梗阻性胆管损伤,强烈刺激胆管反应,可作为小鼠胆管反应模型7.暴露于胆道毒物3,5 -二氧羰基-1,4-二氢碰撞碱(DDC)或α-萘基异硫氰酸酯(ANIT)也会深刻诱导啮齿动物的导管反应,导致胆汁淤积8,9.然而,介导导管反应的胆管细胞内在通路仍然知之甚少。
NF-κ b诱导激酶(NIK),也称为MAP3K14,由一组细胞因子激活,是激活非典型NF-κB2通路所必需的10.TNF-like weak inducer of apoptosis (TWEAK)是一种刺激nik的细胞因子,有趣的是,TWEAK通过其受体Fn14刺激胆管细胞增殖11,12,13.NIK磷酸化并激活IκB激酶-α (IKKα),也称为Chuk14,15,16,17.IKKα反过来磷酸化NF-κB2前体p100,导致p100的蛋白水解裂解产生成熟的转录因子p5218,19.此外,NIK还激活MAPK通路并抑制JAK2/STAT3通路20.,21.在功能上,淋巴样NIK对免疫系统发育和免疫反应起着关键的调节作用10,22.全球尼克基因敲除会损害淋巴结和胸腺的发育23.髓质胸腺上皮特异性尼克基因敲除阻断小鼠胸腺髓质发育,导致致命性自身免疫性肝炎24.我们报道肝脏NIK在肥胖和慢性肝病中上调15,23.糖尿病患者肝脏NIK增加肝糖生成、高血糖和葡萄糖耐受不良14.肝脏NIK的异常激活也通过损害(至少部分地)再生肝细胞增殖,加剧肝毒素诱导的小鼠肝衰竭21,25.然而,在本研究之前,胆道NIK尚未被探索。在此,我们发现在小鼠和人类中,在胆汁淤积性肝损伤的胆管细胞中,NIK被强烈上调。小鼠胆管细胞特异性尼克击倒(尼克ΔK19)对DDC-、ANIT-和bdl诱导的导管反应、肝脏炎症和纤维化具有耐药性。同样,NIK抑制剂治疗也减轻了ddc诱导的导管反应、肝损伤和肝纤维化。我们证明了胆道NIK对胆管细胞可溶性因子(称为胆管因子)的分泌有关键的调节作用,这些可溶性因子刺激肝巨噬细胞和肝星状细胞(hsc)并塑造肝脏微环境。我们的研究结果揭示了胆道NIK是一种以前未被认识到的分子驱动因素,可用于导管反应、肝损伤、炎症和纤维化,并提出了NIK抑制剂可能具有治疗肝脏疾病的潜力的可能性。
结果
胆道NIK在患有慢性肝病的小鼠和人类中上调
为了检测人类胆道NIK,我们用NIK和胆管细胞标记物角蛋白-19 (K19)的抗体对肝脏切片进行了免疫染色。为了验证抗nik抗体,我们将野生型(尼克+/+),以增加肝脏NIK水平,并使用global尼克击倒(尼克−/−)小鼠作为阴性对照。肝切片采用抗nik抗体染色。NIK-expressing(尼克+)细胞很容易被检测到尼克+/+老鼠但不在尼克−/−小鼠(补充图)1)。在人类样本中,我们观察到一些NIK+细胞和K19+健康肝脏中的胆管细胞(对照)(图2)。1)。K19的编号+细胞,尼克+细胞和K19+尼克+细胞(表达nik的胆管细胞)在原发性胆道炎(PBC)、原发性硬化性胆管炎(PSC)、乙型肝炎病毒(HBV)、丙型肝炎病毒(HCV)和酒精性肝硬化中明显升高(图2)。1 a, B)。表达NIK的胆管细胞可能被低估,因为抗NIK抗体可能无法检测到相对较低水平的NIK。

一个,B人肝切片用NIK和K19抗体染色。一个代表图像。标尺:200 μm。B尼克+、K19+,以及NIK+K19+细胞计数并归一化为总细胞数。控制:n= 3名受试者,PBC:n= 3个科目,PSC:n= 3例受试者,HBV:n= 5名受试者,HCV:n= 3名受试者,酒精:n= 3名受试者。C,DC57BL/6J雄性小鼠分别饲喂鼠粮和DDC日粮4周。C肝脏切片用NIK和K19抗体染色。尼克+、K19+,以及NIK+K19+细胞计数并归一化为总细胞数。食物:n= 3只小鼠,DDC:n= 3只老鼠。标尺:200 μm。D采用qPCR检测肝脏NIK的表达(归一化至18s水平)。食物:n= 4只小鼠,DDC:n= 6只老鼠。任意单位。E,FC57BL/6J男性接受BDL或假手术治疗7 d。E肝脏切片用NIK和K19抗体染色。尼克+、K19+,以及NIK+K19+细胞计数并归一化为总细胞数(n=每组3只小鼠)。标尺:200 μm。F用qPCR检测肝脏NIK的表达(归一化至36B4水平;n=每组4只)。数据以平均值±SEM表示。*p< 0.05,双尾学生t以及。源数据作为源数据文件提供。
为了确定胆汁淤积小鼠胆道NIK是否上调,我们将C57BL/6J小鼠置于DDC饮食4周,并用NIK和K19抗体染色肝脏切片。我们观察到一些NIK+细胞和K19+小鼠的胆管细胞(图2)1 c)。DDC饲喂显著增加了NIK的数量+细胞和K19+cholangiocytes;重要的是,大多数胆管细胞表达NIK (NIK)+K19+)(图。1 c)。值得一提的是,NIK+K19+胆管细胞可能被低估,因为抗NIK抗体可能无法识别相对较低水平的NIK。与这些数据一致,DDC喂养显著增加了肝脏尼克mRNA丰度(图2)1 d)。ANIT喂养3周(补充图)1 b, C)或BDL治疗7天(图2)。1 e, F)也显著增加了NIK+K19+胆管细胞数量,肝脏尼克mRNA水平和导管反应。因此,胆道NIK上调是胆道损伤的标志,无论病因如何。
为了测试胆管细胞毒物是否直接刺激胆道NIK,我们分离了原代胆管细胞尼克/ f用E1A慢病毒载体永生化小鼠,并通过一系列稀释纯化单株。这些细胞系表达胆管细胞谱系基因,包括K19,雌性生殖道,Hnf1β(补充图。2 a, B),被认为是胆管细胞系。我们用DDC或ANIT治疗胆管细胞。DDC和ANIT均显著升高尼克mRNA丰度(补充图)2摄氏度)。内源性NIK无法被抗NIK抗体检测,因此我们用NIK腺病毒载体转导胆管细胞。DDC或ANIT显著提高了NIK蛋白水平。二维)。为了评估NIK蛋白的降解,我们先用DDC或ANIT预处理胆管细胞,然后用蛋白翻译抑制剂环己亚胺处理。DDC或ANIT预处理大大提高了NIK的稳定性。2 e, F)。我们从DDC或anti处理的细胞(1/2量的载体处理细胞)加载较少的裂解物,以避免NIK超载。为了扩展这些观察结果,我们研究了NIK的泛素E3连接酶复合物。Traf2和Traf3结合NIK并将其招募到cIAP1和cIAP2,而cIAP1/2又催化NIK泛素化10.DDC治疗显著降低了cIAP1和cIAP2,但增加了胆管细胞培养中Traf2和Traf3的水平(补充图)。2 g)。为了评估体内的cIAP1/2,我们通过免疫印迹法测定了肝脏提取物中Traf2、Traf3、cIAP1和cIAP2的水平。DDC饲喂显著降低肝脏cIAP1和cIAP2水平,提高肝脏Traf2和Traf3水平(补充图)。2 h)。为了进一步证实肝脏cIAP1和cIAP2的下调,我们用不同来源的cIAP1和cIAP2抗体对肝脏提取物进行免疫印迹。ddc处理小鼠肝脏cIAP1和cIAP2被深度下调(补充图)。2我)。动物饲养同样降低肝脏中cIAP1/2水平(补充图2)。2 j)。cIAP1/2的下调至少部分解释了NIK稳定性的增加,也许,Traf2/3的上调是对cIAP1/2缺乏的次要反应。综上所述,这些结果表明胆道损伤增加了胆管细胞NIK的表达和稳定性。
胆管细胞特异性缺失尼克抑制DDC-和anti诱导的导管反应
我们试图在体内探索胆道NIK。我们产生了他莫昔芬诱导的胆管细胞特异性尼克击倒(尼克ΔK19杂交老鼠尼克/ f老鼠K19-CreERT驱动程序。尼克/ f和K19-CreERT老鼠以前也被描述过26,27.因为尼克和K19-CreERT基因位于同一条染色体上,尼克f / +; K19-CreERT+/−重组在生殖细胞减数分裂中发生的频率很低。尽管如此,我们得到了尼克/ f; K19-CreERT+/−老鼠。尼克/ f; K19-CreERT+/−小鼠(7-10周龄)用他莫昔芬治疗尼克特别是在胆管细胞中,产生尼克ΔK19老鼠。验证胆管细胞特异性缺失尼克我们制备了原代胆管细胞和肝细胞尼克/ f; K19-CreERT老鼠,用他莫昔芬治疗,并测量了被破坏的尼克等位基因(尼克−/−)进行基因分型。的尼克−/−在他莫昔芬处理的胆管细胞中检测到等位基因,但在他莫昔芬处理的肝细胞或载体处理的胆管细胞中未检测到等位基因。3)。我们把尼克ΔK19和对照组小鼠(他莫昔芬治疗后2周),DDC饮食再持续4周。我们使用了2个对照组。尼克/ f; K19-CreERT+/−小鼠用橄榄油培养液作为对照(以下简称橄榄油培养液)尼克/ f)。作为额外的控制,尼克/ f小鼠用他莫昔芬治疗2周,然后用DDC喂养4周。在他莫昔芬处理和橄榄油处理之间,ddc引起的肝损伤水平相当尼克/ f小鼠(补充图)3 b),表明他莫昔芬单独治疗不会影响肝脏对DDC的反应。考虑到他莫昔芬治疗尼克/ f小鼠与给药小鼠无法区分尼克/ f; K19-CreERT小鼠,它们没有被纳入接下来的实验。
DDC喂养降低了两者的体重尼克ΔK19和尼克/ f同窝的同伴,但是到了更高的层次尼克/ f小鼠(补充图)3 c)。为了粗略估计胆道生长,我们使用胶原酶解剖肝内胆管。DDC喂养增加了两者的总胆管重量尼克ΔK19和尼克/ f一窝一窝的伴侣,但在尼克/ f小鼠(补充图)3 d)。为了直接检查导管反应,我们用K19和NIK抗体对肝脏切片进行了联合染色。正如预期的那样,DDC喂养引起了剧烈的导管反应和NIK的膨胀+K19+野生型胆管细胞尼克/ f老鼠(图。2 a, B)。K19+胆管细胞数量急剧减少尼克ΔK19老鼠(图。2)。肝脏尼克+K19+细胞在尼克ΔK19老鼠(图。2 a, B),证实尼克在胆管细胞中特异性缺失。为了进一步验证尼克删除在尼克ΔK19用抗nf -κB2抗体对小鼠肝提取物进行免疫印迹。DDC饲喂显著提高大鼠NF-κB2 p100水平和p100向p52转化(NIK激活指数)尼克/ fp52水平显著降低尼克ΔK19老鼠(图。2摄氏度)。为了证实胆道NIK缺乏阻断ddc诱导的小管反应,我们用抗k19抗体对肝切片进行免疫染色。我们观察到一些K19+饲养的胆管细胞尼克/ f和尼克ΔK19老鼠;重要的是,DDC饲喂提高了K19+胆管细胞数量急剧增加尼克/ f相对于尼克ΔK19老鼠(图。2 d, E)。肝K19mRNA水平也显著高于尼克/ f比在尼克ΔK19老鼠(图。2 f)。尼克+HNF4α+在正常喂养的小鼠中几乎检测不到肝细胞,在DDC喂养后肝细胞略有增加(补充图)。3 e, F)。与尼克+K19+cholangiocytes,尼克+HNF4α+肝细胞可比较尼克/ f和尼克ΔK19小鼠(补充图)3 f)。为了扩展这些发现,我们放置尼克ΔK19和尼克/ f在3周的时间里,一窝一窝的幼崽都在吃ANIT饮食。ANIT,像DDC一样,诱导了胆管细胞的明显扩张尼克/ f老鼠;重要的是,anit诱导的导管反应也显著减弱尼克ΔK19与老鼠相比尼克/ f老鼠(图。2 g及补充图3 g)。这些结果揭示了NIK是胆管反应不可或缺的胆管细胞内在诱导剂。

一个- - - - - -F尼克ΔK19和尼克/ f雄性小鼠分别饲喂鼠粮和DDC日粮4周。一个,B肝脏切片检测NIK和K19抗体。尼克+K19+细胞计数并归一化为K19+细胞(n=每组3只小鼠)。标尺:200 μm。C肝提取物用NF-κB2和β-肌动蛋白抗体进行免疫印迹(每条通道代表一只小鼠;每组4只)。D,E肝切片用抗k19抗体免疫染色。D代表图像。E计数K19胆管细胞并归一化为总细胞数。食物:尼克/ f:n= 4只;尼克ΔK19:n= 4只小鼠;监护系统:尼克/ f:n= 7只;尼克ΔK19:n= 8只小鼠。标尺:200 μm。F肝脏K19表达(归一化至18s水平)。任意单位。食物:尼克/ f:n= 4只;尼克ΔK19:n= 4只小鼠;监护系统:尼克/ f:n= 6只小鼠;尼克ΔK19:n= 6只老鼠。G尼克ΔK19(n= 6只小鼠)和尼克/ f雄性(n = 7只)饲喂ANIT日粮3周。肝切片用抗k19抗体染色。计数K19胆管细胞并与肝细胞总数归一化。数据以平均值±SEM表示。*p< 0.05, 2-way ANOVAB,E,F还有双尾学生的t以及G.源数据作为源数据文件提供。
胆道NIK促进胆管细胞增殖,抑制胆管细胞死亡
我们开始询问尼克诱导的导管反应的细胞机制。我们把尼克/ f和尼克ΔK19DDC饮食4周。肝切片用K19和Ki67(增殖标志物)或K19和cleaved caspase-3(凋亡标志物)抗体共免疫染色。增殖Ki67的数量+K19+胆管细胞明显降低尼克ΔK19比在尼克/ f老鼠;相反,凋亡的caspase -3+K19+胆管细胞明显增高尼克ΔK19老鼠(图。3)。使用TUNEL试验,我们证实胆管细胞死亡在尼克ΔK19相对于尼克/ fDDC后的窝仔饮食(补充图)。4)。值得注意的是,HNF4α的增殖+肝细胞和F4/80+Kupffer细胞和肝细胞死亡在两组之间具有可比性尼克ΔK19和尼克/ f小鼠(补充图)4)。我们还检测了anit喂养小鼠(3周)的胆管细胞增殖和死亡情况。胆管细胞增殖(Ki67+K19+而胆管细胞死亡率(caspase -3+K19+)显著高于尼克ΔK19老鼠比老鼠多尼克/ f老鼠(图。3 b)。

尼克ΔK19和尼克/ f雄性小鼠饲喂DDC日粮4周(一个)或ANIT饮食3周(B)。肝切片用K19和Ki67抗体或裂解caspase-3共免疫染色。增殖(Ki67+K19+)和凋亡(Casp3+K19+)胆管细胞计数并归一化为总K19+cholangiocytes。如DDC / Ki67:尼克/ f:n= 7只;尼克ΔK19:n= 8只小鼠;如DDC / Casp3:尼克/ f:n= 4只;尼克ΔK19:n= 4只小鼠;雕像/ Ki67:尼克/ f:n= 7只;尼克ΔK19:n= 6只小鼠;雕像/ Casp3:尼克/ f:n= 7只;尼克ΔK19:n= 6只老鼠。标尺:200 μm。C尼克+/+和尼克−/−用FBS或TWEAK (20 ng/ml)处理胆管细胞24 h。计数胆管细胞并归一化为初始数目(n= 3组)。D尼克+/+和尼克−/−在BrdU存在的情况下,用10% FBS或TWEAK (10 ng/ml)刺激胆管细胞14小时,然后用抗BrdU抗体进行免疫染色。BrdU+计数胆管细胞并归一化为总胆管细胞。尼克−/−/10% FBS组:n= 4次重复,其他组:n= 5次重复。E尼克+/+和尼克−/−将胆管细胞去除FBS或用棕榈酸酯(PA)处理24小时,然后进行TUNEL检测。TUNEL+细胞计数并归一化为总细胞数(n=每组重复3次)。F尼克+/+和尼克−/−用wortmannin (Wort)或LY294002 (LY)预处理胆管细胞,然后在BrdU存在下用TWEAK刺激胆管细胞。BrdU+计数胆管细胞并归一化为总胆管细胞(n=每组重复3次)。数据以平均值±SEM表示。*p< 0.05,双尾学生t以及一个,B,E,F或单因素方差分析C,D.源数据作为源数据文件提供。
为了测试NIK细胞是否能自主调节胆管细胞的增殖和存活,我们制备了尼克-胆管细胞系缺陷。我们制备了胆管细胞系尼克/ f小鼠(补充图)2 a, B)。尼克/ f胆管细胞(以下简称胆管细胞尼克+/+)用Cre腺病毒载体转导进行删除尼克(尼克−/−),然后通过反复传代去除腺病毒载体。确认尼克我们刺激了缺失尼克+/+和尼克−/−胆管细胞有TWEAK。调整刺激的NF-κB2 p52的产生尼克+/+但不是尼克−/−胆管细胞(补充图)5)。为了检验增殖,我们刺激尼克+/+和尼克−/−用TWEAK或胎牛血清(FBS)检测胆管细胞,并计数细胞数量。调整增加尼克+/+但不是尼克−/−胆管细胞数量(图2)3 c)。的边后卫增加尼克+/+相对于胆管细胞水平更高尼克−/−cholangiocytes(无花果。3 c)。我们用BrdU掺入试验验证了这些结果。brdu阳性细胞在尼克+/+比在尼克−/−TWEAK或FBS刺激后的胆管细胞培养(图。3 d及补充图5 b)。为了诱导细胞凋亡,将胆管细胞与血清分离24小时或用棕榈酸处理,用TUNEL法测定细胞死亡。棕榈酸酯有诱导胆管细胞死亡的报道28.血清饥饿或棕榈酸盐治疗增加TUNEL+细胞中含量显著升高尼克−/−比在尼克+/+胆管细胞培养(图2)3 e及补充图5度)。血清剥夺使细胞活力降低到较高水平尼克−/−比在尼克+/+胆管细胞培养,通过MTT测定(补充图)。5 d)。这些结果表明,胆道NIK通过促进胆管细胞增殖和抑制其死亡来促进胆管反应。
胆道NIK消融可减轻DDC和anti诱导的肝损伤、炎症和纤维化
我们接下来试图研究导管反应的病理结果。体重、血浆丙氨酸转氨酶(ALT)、碱性磷酸酶(ALP)、胆红素均正常尼克ΔK19鼠粮小鼠(补充图)6 a、B)。我们把尼克ΔK19和尼克/ f用DDC饮食4周的幼崽。血浆ALT在DDC饮食后一周内升高,随后下降,血浆ALP和胆红素水平(胆道损伤标志物)在DDC饮食后逐渐升高(图2)。4)。重要的是,血浆ALT、ALP和胆红素水平显著降低尼克ΔK19比在尼克/ fDDC后小鼠饮食(图2)4)。组织学检查显示,DDC喂养引起严重的肝损伤和炎症;引人注目的是,肝损伤和免疫细胞浸润显著降低尼克ΔK19比在尼克/ f老鼠(图。4 b)。肝脏CD11b+巨噬细胞和Gr-1+髓样细胞明显减少尼克ΔK19比在尼克/ f老鼠(图。4 b, C)。尼克+CD11b+细胞具有可比性尼克ΔK19和尼克/ fDDC日粮小鼠(补充图)3 f)。肝CD4和CD8 T细胞稍低尼克ΔK19小鼠(补充图)4 b)。为了证实这些结果,我们放置尼克ΔK19和尼克/ f在3周的时间里,一窝一窝的幼崽都在吃ANIT饮食。饲料喂养使血浆ALT、ALP和胆红素显著升高尼克/ f老鼠比老鼠多尼克ΔK19小鼠(补充图)7一个)。尼克ΔK19小鼠对anit诱导的肝损伤和炎症也有明显的抵抗力(图2)。4 d, E及补充图7 b)。

一个- - - - - -C尼克ΔK19和尼克/ f雄性小鼠饲喂DDC日粮4周。一个血浆ALT、ALP和总胆红素水平。尼克/ f:n= 8只;尼克ΔK19:n= 9只老鼠。B,C肝切片用H&E或CD11b、Gr-1抗体染色。B代表图像。标尺:200 μm。CCD11b (n=每组3只小鼠)和Gr-1细胞(n=每组3只),并归一化为总肝细胞数(n=每组3-4只小鼠)。D,E尼克ΔK19和尼克/ f雄性小鼠饲喂ANIT日粮3周。D肝切片代表性的H&E染色。标尺:200 μm。E肝切片用抗cd11b抗体染色。计数CD11b细胞并归一化为肝细胞总数。尼克/ f:n= 7只;尼克ΔK19:n= 6只老鼠。数据以平均值±SEM表示。*p< 0.05, 2-way方差分析(一个)和双尾学生的t以及(C,E)。源数据作为源数据文件提供。
我们通过肝切片天狼星红染色和测定肝脏羟脯氨酸含量来检测肝纤维化,并检测表达α-平滑肌肌动蛋白(αSMA)的肝干细胞与肝纤维化有关。天狼星红阳性纤维和αSMA+鼠粮中几乎检测不到造血干细胞,DDC喂养后造血干细胞显著增加(图2)。5)。天狼星红区,羟脯氨酸含量,αSMA+的HSC数量明显降低尼克ΔK19比在尼克/ fDDC组小鼠(图2)5 a、B)。肝提取物αSMA水平显著降低尼克ΔK19比在尼克/ fDDC饲喂后小鼠(图2)5度)。为了确定性别是否影响表型,我们放置尼克ΔK19和尼克/ f雌性小鼠给予DDC饮食4周。尼克ΔK19雌性和雄性一样,对ddc诱导的导管反应、肝损伤、炎症和纤维化具有抗性。8)。为了进一步验证这些发现,我们放置尼克/ f和尼克ΔK19给老鼠喂食3周的ANIT饮食。ANIT强烈刺激αSMA+HSC活化与肝纤维化的关系尼克/ f老鼠;HSC活化和肝纤维化在很大程度上被阻断尼克ΔK19老鼠(图。5 d)。这些结果首次表明,胆管细胞内生性NIK可能通过诱导导管反应促进肝损伤、炎症和纤维化。

一个,B尼克ΔK19和尼克/ f雄性小鼠饲喂DDC日粮4周。肝切片采用天狼星红/快绿染色或αSMA抗体染色。一个代表图像。标尺:200 μm。B小天狼星红(尼克/ f:n= 6只小鼠;尼克ΔK19:n= 7只小鼠)和αSMA HSC (n=每组4只小鼠)面积测量并归一化为总面积。测定肝脏羟脯氨酸水平,并与肝脏重量(尼克/ f:n= 6只小鼠;尼克ΔK19:n= 8只小鼠)。C小鼠分别饲喂鼠粮和DDC日粮4周。肝提取物用抗α - sma和p85(负载对照)抗体进行免疫印迹。每条车道代表一只动物(n=每组3只小鼠)。D尼克ΔK19(n= 6只小鼠)和尼克/ f(n(7只)雄性小鼠饲喂ANIT日粮3周。肝切片用天狼星红/快绿或抗α sma染色。标尺:200 μm。天狼星红和αSMA区域归一化为总面积。数据以平均值±SEM表示。*p< 0.05,双尾学生t以及。源数据作为源数据文件提供。
尼克ΔK19小鼠对bdl诱导的导管反应和肝损伤具有抗性
为了测试胆道NIK是否提供了导管反应和肝脏疾病的共同途径,我们进行了7天的BDL尼克ΔK19和尼克/ f老鼠。血浆ALT、ALP和胆红素水平在假药组之间比较尼克ΔK19和尼克/ f小鼠(补充图)9)。BDL使血浆ALT、ALP和胆红素水平显著升高尼克/ f比在尼克ΔK19同窝出生(无花果。6)。bdl诱导的肝损伤、小管反应(K19染色)、肝脏炎症(H&E、CD11b和Gr-1染色)和纤维化(天狼星红和αSMA染色)尼克/ f老鼠(图。6 b),尼克ΔK19小鼠对bdl诱导的肝损伤、小管反应、炎症和纤维化具有抗性(图2)。6 b, C)。胆管细胞增殖(K19+Ki67+胆管细胞凋亡(K19+caspase-3+的比例更高。尼克ΔK19相对于尼克/ fBDL后小鼠(图2)6 b及补充图9 b)。总之,这些发现表明,胆道NIK整合了来自各种胆道损伤的信息,作为一个汇合点,诱导了导管反应、肝损伤、炎症和纤维化。

行BDL尼克ΔK19和尼克/ f7天后对雄性小鼠进行肝脏分析。一个血浆ALT、ALP和总胆红素水平(n=每组6只)。B,C肝脏切片用指定抗体染色。B代表图像。标尺:200 μm。C对单个亚群进行计数,并将其归一化为总肝细胞。天狼星红色区域被归一化为总视野区域(n=每组3只小鼠)。肝脏羟脯氨酸含量与肝脏重量(尼克/ f:n= 5只小鼠;尼克ΔK19:n= 6只小鼠)。K19:n每组3只;Casp3+K19+:n每组3只;Ki67+K19+:n每组3只;Gr-1:n每组4只;CD11b:n每组4只;αSMA:n每组4只。数据以平均值±SEM表示。*p< 0.05,双尾学生t以及(C)和双因素方差分析(一个)。源数据作为源数据文件提供。
胆道NIK促进促炎症和促纤维化胆管因子的分泌
接下来,我们着手研究胆道nik引发的导管反应促进肝脏炎症和纤维化的机制。我们推测NIK可能刺激胆管因子(胆管细胞来源的介质)的分泌,从而激活肝脏免疫细胞和造血干细胞,增加肝脏炎症和纤维化。我们刺激尼克+/+和尼克−/−用TWEAK激活NIK的胆管细胞培养和测量各种已知参与肝脏炎症和纤维化的细胞因子的表达。NIK消融显著降低Il-1β、Il-4、Il-6、iNos、Tnfα、Mcp1(也称为Ccl2)和Tgfβ1的表达尼克−/−胆管细胞相对于尼克+/+cholangiocytes(无花果。7一个)。为了测试nik上调胆管因子是否会促进肝脏炎症,我们分离小鼠骨髓源性巨噬细胞(bmdm)并与两者共培养尼克+/+或尼克−/−正如我们之前所描述的那样,transwell中的胆管细胞15.用TWEAK预处理胆管细胞激活NIK。通过测量炎症介质的表达来评估BMDM活性。Il-1β、Il-6、iNos、Tnfα、Mcp1和Ccl5的表达显著降低尼克−/−胆管细胞共培养bmms相对于尼克+/+胆管细胞-共培养BMDMs(图。7 b)。为了进一步证实胆道NIK刺激激活BMDMs的胆管因子的分泌,我们刺激尼克+/+和尼克−/−用TWEAK增加胆管细胞向生长培养基分泌胆管因子,并从这些细胞制备条件培养基。弹道导弹治疗用尼克+/+或尼克−/−测定胆管细胞条件培养基及其炎症介质的表达。Il-1β、Il-6、iNos、Tnfα、Mcp1和Ccl5的表达显著降低尼克−/−条件介质处理bmms比尼克+/+条件培养基处理的弹道导弹(图。7 c)。为了确定nik上调的胆管因子是否刺激hsc和肝纤维化,我们用尼克+/+或尼克−/−并测量其纤维化相关基因的表达。αSMA、Col1a1、Timp1、Mmp9、Ctgf、Tgfβ1的表达均显著降低尼克−/−比在尼克+/+条件培养基刺激的造血干细胞(图。7 d)。这些结果提供了概念证据,证明胆道NIK能有效刺激胆管因子的分泌,从而激活肝脏免疫细胞和造血干细胞,从而促进肝脏炎症和纤维化。

一个用TWEAK (50 ng/ml)处理胆管细胞培养48 h。采用qPCR检测基因表达,归一化至36B4表达(n=每组重复3次)。BBMDM与twist刺激的胆管细胞共培养24 h,用qPCR检测BMDM基因表达,并归一化至36B4表达(n=每组重复3次)。CBMDMs或D用胆管细胞条件培养基分别处理造血干细胞6 h和24 h。qPCR检测基因表达,归一化至36B4水平(n=每组重复3次)。E胆道损伤(胆道毒素、氧化应激、细胞因子)激活胆道NIK。NIK可能通过Akt通路增强胆管细胞增殖,同时抑制胆管细胞死亡,从而诱导管反应。此外,NIK刺激反应性胆管细胞分泌胆管因子。胆管因子激活肝脏免疫细胞和造血干细胞,促进肝脏炎症、肝纤维化和肝脏疾病进展。数据以平均值±SEM表示。*p< 0.05,双尾学生t以及。
胆道IKKα消融不影响ddc诱导的导管反应和胆汁淤积性肝损伤
为了测试IKKα是否介导NIK在胆管因子中的作用,我们制作了他莫昔芬诱导的胆管细胞特异性的IKKα击倒(IKKαΔK19杂交老鼠IKKα/ f与K19-CreERT驱动程序。IKKα/ f;K19-CreERT+/−小鼠服用他莫昔芬(IKKαΔK19)或车辆(被称为IKKα/ f)。2周后,连续4周饲喂DDC日粮。K19中检测到IKKα+cholangiocytes在IKKα/ f但不是IKKαΔK19免疫染色小鼠(补充图)10),证实胆管细胞特异性IKKα淘汰赛。IKKαΔK19小鼠没有发生表型尼克ΔK19老鼠。血浆ALT、ALP和胆红素水平在两组间无明显差异IKKαΔK19和IKKα/ f小鼠(补充图)10 b)。胆管细胞扩张,αSMA+两组间的HSC数和肝纤维化也具有可比性IKKαΔK19和IKKα/ f小鼠(补充图)10 c)。与胆道NIK不同,ddc诱导的胆管反应和胆汁淤积不需要胆道IKKα。因为IKKαΔK19老鼠藏着K19-CreERT这些数据表明,他莫昔芬激活CreERT不影响ddc诱导的导管反应。因此,IKKαΔK19小鼠也被作为额外的对照尼克ΔK19老鼠。
Akt可促进胆管细胞扩张3.,促使我们测试它是否是NIK通路的下游介质。我们刺激尼克+/+和尼克−/−用TWEAK培养胆管细胞,免疫印迹法检测Akt磷酸化水平。微调刺激Akt磷酸化/激活到更高水平尼克+/+比在尼克−/−细胞(补充图)5 e)。接下来,我们使用BrdU检测阻断Akt激活是否会减弱nik刺激的胆管细胞增殖。培养的胆管细胞用PI 3-激酶抑制剂wortmannin或LY294002预处理,然后用TWEAK刺激。TWEAK使细胞增殖率提高到较高水平尼克+/+胆管细胞相对于尼克−/−cholangiocytes;重要的是,wortmannin或LY29402对Akt的抑制可以消除由微调刺激的细胞增殖尼克+/+cholangiocytes(无花果。3 f)。相反,wortmannin和LY29402没有影响尼克−/−cholangiocyte增殖。因此,NIK至少在一定程度上通过增强Akt的激活来促进胆管细胞的扩张。
NIK抑制剂治疗可改善ddc诱导的导管反应、肝损伤和纤维化
nik选择性抑制剂化合物33 (C33)已被开发用于治疗癌症29.我们评估了C33在肝脏疾病治疗中的治疗潜力。为了验证C33的有效性,我们用NF-κB2/p100和NIK质粒共转染HEK293细胞,然后用C33处理。C33以剂量依赖的方式抑制p100裂解成p52(由NIK刺激)。8)。C33还抑制NIK刺激NF-κB荧光素酶报告活性的能力(图2)。8 b)。为了研究C33在体内的作用,我们将C57BL/6 J雄性小鼠置于DDC饮食3周,同时给予C33 (10 mg/kg体重,每周2次,对照组)。C33处理小鼠的肝脏NF-κB2/p52水平明显低于药物处理小鼠,但未被C33处理消除(补充图2)。11个),说明在实验条件下,C33部分抑制了NIK。C33治疗显著降低血浆ALT、ALP和胆红素水平(图2)。8 c)。K19的编号+胆管细胞、表达髓过氧化物酶(MPO)的中性粒细胞和αSMA+c33处理小鼠的造血干细胞和肝纤维化(天狼星红染色)明显低于载药处理小鼠(图2)。8 d, E)。Cholangiocytes (Ki67+K19+)和HSC (Ki67+αSMA+), c33处理小鼠的增殖率明显低于载药处理小鼠(补充图2)。11 b, C)。相反,肝细胞(HNF4α+Ki67+)和Kupffer细胞(Ki67+F4/80+)增殖率在C33组和载药组之间具有可比性。这些结果表明,药理抑制NIK能够减轻小鼠的导管反应、肝脏炎症和纤维化。

一个用NF-κB2和NIK表达质粒共转染HEK293细胞,并用C33处理。用NF-κB2和NIK抗体对细胞提取物进行免疫印迹。B用NF-κB荧光素酶报告基因和NIK表达质粒共转染HEK293细胞,并用C33处理。在C33处理48 h后评估荧光素酶活性(n=每组重复3次)。C- - - - - -EC57BL/ 6j雄性用DDC和C33治疗3周。C血浆ALT、ALP和总胆红素水平(n=每组8只)。D,E肝切片用指定抗体进行免疫染色。D代表性肝脏图像。标尺:200 μm。EK19+和MPO+细胞计数并归一化为肝细胞总数。天狼星红和αSMA HSC区域归一化为总面积。K19、MPO、αSMA:n每组8只,天狼星红;n每组4只。数据以平均值±SEM表示。*p< 0.05,双尾学生t以及(E)及单程(B)或双向(C方差分析。源数据作为源数据文件提供。
讨论
导管反应常见于慢性肝病,包括胆汁淤积、酒精性肝病和非酒精性脂肪性肝炎(NASH)。导管反应与预后不良相关,是肝内胆管癌的重要危险因素3.,6,30..胆道损伤与异常的肝脏微环境相一致,驱动胆管细胞扩张和导管反应。在这项研究中,我们发现胆管细胞内生性NIK是连接胆管损伤、肝损伤、炎症和纤维化的分子桥梁。我们证明DDC、ANIT或BDL治疗通过增加其表达和稳定性显著上调小鼠胆道NIK。一致地,DDC和ANIT直接增加了胆管细胞培养中NIK的表达和稳定性。在野生型小鼠中,DDC、ANIT或BDL如预期的那样诱导了导管反应、肝损伤、炎症和纤维化。引人注目的是,胆管细胞特异性消融NIK可阻断DDC-、ANIT-和bdl诱导的导管反应、肝损伤和纤维化尼克ΔK19老鼠。同样,C33对NIK的药理抑制也减轻了ddc诱导的小鼠小管反应、肝损伤、炎症和纤维化。胆管细胞增殖较低,而死亡率较高尼克ΔK19相对于尼克/ f小鼠经过DDC, ANIT或BDL治疗后,这可能解释了尼克ΔK19小鼠对胆管细胞扩张及小管反应。NIK可被细胞因子、细胞内应激和细胞损伤因子激活14,21,提出了胆道NIK可能整合广泛的胆道损伤相关信号以促进导管反应、肝损伤、炎症和纤维化的可能性(图2)。7 e)。值得注意的是,胆道NIK在PBC、PSC、HBV、HCV或酒精性肝病患者中显著上调,这增加了一种有趣的可能性,即类似的胆道NIK/导管反应/肝脏炎症/肝纤维化级联反应也可能在人类肝病进展中起作用。
考虑到IKKα在NIK下游激活非典型NF-κB2通路,这是出乎意料的IKKαΔK19小鼠没有发生表型尼克ΔK19老鼠。与这些发现一致,消融胆管细胞relB (NF-κB2的伴侣)也不会影响ddc诱导的导管反应31.因此,胆道NIK似乎通过IKKα/NF-κ b2独立的机制促进导管反应。我们观察到TWEAK刺激了NF-κB2和Akt通路尼克+/+cholangiocytes。正如预期的那样,NIK缺乏完全阻断了tweak刺激的非典型NF-κB2通路的激活尼克−/−cholangiocytes。NIK缺乏也显著减弱了tweak刺激下Akt的磷酸化尼克−/−表明TWEAK或相关细胞因子刺激了胆管细胞的NIK/Akt通路。已知TWEAK可刺激胆道生长和扩张,而消融TWEAK受体Fn14可抑制管状反应11.Akt也被认为介导胆道增生3..药理抑制PI 3-激酶/Akt通路可阻断tweak刺激的细胞增殖尼克+/+胆管细胞,但不是尼克−/−这表明胆道NIK/Akt通路至少部分介导了胆管反应(图2)。7 e)。
导管反应长期以来被认为与慢性肝病有关,但其对肝病进展的贡献仍不清楚。电阻的尼克ΔK19小鼠对DDC、ANIT和bdl诱导的肝脏炎症和纤维化的影响,有力地证明了胆道nik引发的导管反应是肝脏炎症和纤维化的一个原因。在导管反应中,反应性胆管细胞获得促炎和促纤维化特性3.,32.我们观察到炎症介质(Il-1β、Il-4、Il-6、iNos、Tnfα和Mcp1)和促纤维化基因(Tgfβ1)的表达明显降低尼克−/−胆管细胞相对于尼克+/+cholangiocytes。与这些结果一致,巨噬细胞被激活到更高的水平尼克+/+胆管细胞相对于尼克−/−胆管细胞,并通过刺激到更高的水平尼克+/+胆管细胞条件培养基相对于尼克−/−cholangiocyte-conditioned媒介。同样的,尼克+/+胆管细胞条件培养基,含有胆管因子,比尼克−/−胆管细胞条件培养基刺激HSC活化及其促纤维化基因的表达(αSMA,Col1a1,Mmp9,Ctgf,转化生长因子β1)。基于这些发现,我们很想假设在慢性肝病中上调的胆道NIK直接刺激胆管因子的表达和分泌,进而激活肝脏免疫细胞和造血干细胞(图2)。7 e)。胆道NIK/胆管因子/免疫细胞轴和胆道NIK/胆管因子/HSC轴驱动肝脏炎症和纤维化的发病机制。如上所述,胆道NIK诱导导管反应,可以想象NIK的扩张+胆管细胞进一步增加胆管因子的分泌,形成致病性肝脏微环境并加剧肝脏疾病的进展。
C33治疗显著减轻了ddc诱导的导管反应、肝脏炎症和纤维化,这增加了NIK抑制剂作为抗肝病候选药物的有趣可能性。C33不如胆道基因缺失有效尼克预防肝脏疾病,因为在实验条件下,它部分抑制了肝脏NIK。值得注意的是,这项研究有局限性。我们不能排除C33在体内除胆管细胞外可能靶向其他细胞类型的可能性。全局或胸腺上皮细胞特异性缺失尼克损害小鼠胸腺发育和中枢T细胞耐受性23,24这引起了人们对NIK抑制剂安全性的担忧。然而,人类胸腺在成人中是退化的,这反驳了NIK抑制剂可能损害成人T细胞适应性免疫的可能性。介导胆管细胞与巨噬细胞之间以及胆管细胞与造血干细胞之间的串扰的单个胆管因子的身份仍有待确定。胆道NIK/Akt通路对导管反应的作用有待于体内验证。需要进一步加强人肝活检,以巩固胆道NIK在人肝脏疾病中的结论。这些问题指出了未来的研究方向。
方法
动物
尼克/ f,IKKα/ f和K19-CreERT小鼠(C57BL/6J背景),如前所述26,27,33.尼克和K19都在11号染色体上的尼克f / +; K19-CreERT−/+基因型是通过生殖细胞减数分裂(低频率)的同源重组产生的。尼克/ f; K19-CreERT老鼠和IKKα/ f; K19-CreERT小鼠于7-8周龄给予他莫昔芬(5 mg/只,2次,间隔4天,每次1次)产生尼克ΔK19和IKKαΔK19老鼠,分别。以橄榄油为对照。尼克/ f小鼠注射他莫昔芬作为额外的对照。小鼠被置于12小时的明暗循环和25小时0在密歇根大学(ULAM)实验动物医学单元的环境温度C和饲料随意食物饮食(9%的热量是脂肪;TestDiet,圣路易斯,密苏里州),DDC饮食,或ANIT饮食。
伦理语句
动物研究符合所有相关民族法规。动物实验遵循密歇根大学机构动物护理和使用委员会(IACUC)批准的方案进行。
人体肝脏活检
去鉴定的人类肝脏标本由Suthat Liangpunsakul博士根据印第安纳-普渡大学印第安纳波利斯分校irb批准的方案提供,由Anna S. Lok博士根据密歇根大学irb批准的方案提供。本研究的设计和行为得到了印第安纳-普渡大学印第安纳波利斯分校和密歇根大学IRB委员会的授权,并遵守了有关使用人类研究参与者的所有相关规定。患者提供知情同意。这项研究是按照《赫尔辛基宣言》规定的标准进行的。
DDC饮食和ANIT饮食
他莫昔芬治疗后2周(清除胆道尼克或IKKα),尼克ΔK19,IKKαΔK19对照组小鼠分别饲喂DDC饲粮4周和ANIT饲粮3周。将0.1%的DDC (Sigma-Aldrich, 137030)或0.08%的ANIT (Sigma-Aldrich, 18951)添加到饲料饲料粉(LabDiet 5001)中,配制成DDC和ANIT饲料。
BDL
尼克ΔK19和尼克/ f小鼠(9-10周龄)异氟醚麻醉,开腹正中(~ 1cm)显露胆总管。用5-0丝线在胆总管上做两个结。胆管在两个结之间切开。腹膜重新排列。皮肤和下面的肌肉层分别用5-0丝线缝合。
复方33处理
C57BL/6J雄性小鼠(8周)在饲喂DDC日粮的同时,以10 mg/kg体重剂量给药NIK抑制剂化合物33(中国科学院上海药物研究所)(每周2次,ig),连续3周。化合物33在10% PEG400(聚乙二醇,分子量400,Sigma #P3265)和3% Cremophor EL (Sigma #C5135)的载体中制备。分析体重、血液化学和肝脏病理。
血液化学
从尾静脉采集血样。血浆ALT、ALP和胆红素水平使用商业试剂盒(Pointe Scientific Inc., Canton, MI;ALT: A7526625;高山:A7516150;胆红素:B7576120)。
羟脯氨酸测定
肝脏样品用6盐酸匀浆,100℃水解18 h, 9391xg离心5 min。上清液在高速真空中干燥,溶解于H2然后用10n氢氧化钠中和。样品在氯胺- t溶液(60 mM氯胺- t (Sigma, 857319), 20 mM柠檬酸盐,50 mM醋酸盐,pH 6.5)中室温孵育25分钟,然后在65°C的埃利希溶液(Sigma, 038910)中再孵育20分钟。羟脯氨酸含量采用贝克曼库尔特AD 340平板阅读器(570 nm)测量,并与肝脏重量归一化。
肝切片染色
肝脏石蜡切片用0.1%天狼星红(Sigma, 365548)和0.1%快绿(Sigma, F7252)(饱和苦味酸)染色。肝脏冷冻切片使用Leica cryostat (Leica Biosystems Nussloch GmbH, Nussloch, Germany),用4%多聚脲固定30分钟,用5%正常山羊血清(Life Technologies)加1%牛血清阻断3小时,与指定抗体在4°C下孵育过夜。抗体列于补充表1.使用原位细胞死亡检测试剂盒(Roche Diagnostics, Indianapolis, In, 11684817910)用TUNEL试剂对肝脏冷冻切片进行染色。
免疫印迹
细胞或组织在L-RIPA裂解缓冲液(50 mM Tris, pH 7.5, 1% Nonidet P-40, 150 mM NaCl, 2 mM EGTA, 1 mM Na)中均质3.签证官4, 100mm NaF, 10mm Na4P2O7,苄脒1 mM, 10 μg ml−1抑肽素,10 μg ml−1白细胞介素,1毫米苯甲基磺酰氟)。用SDS-PAGE分离蛋白,用指定抗体进行免疫印迹。抗体列于补充表1.HEK293细胞系来自ATCC (CRL-1573)。
胆道树的分离和胆管细胞系的产生
尼克/ f雄性(9周)用异氟醚麻醉,肝脏灌注II型胶原酶(Worthington Biochem, Lakewood, NJ)。分离胆管树,用PBS广泛洗涤,称重,切碎成~ 1mm块,在添加10%胎牛血清和抗生素的DMEM (25 mm葡萄糖)中培养。增生的胆管细胞、门静脉成纤维细胞和造血干细胞在7-8天内从组织块中迁移。收集这些细胞,用E1A慢病毒载体转导,并用嘌呤霉素处理1周,以选择永生化细胞。从单细胞谱系衍生的单株细胞系通过一系列稀释产生。为了鉴定这些细胞系,我们用qPCR方法检测了胆管细胞标志物(K7、K19、Cftr、Hnf1β)和HSC标志物(col-1a1、αSMA)的表达。尼克/ fcholangiocytes (尼克+/+)用Cre腺病毒载体转导进行删除尼克(尼克−/−)。随后通过反复传代(bbb10代)去除腺病毒载体。胆管细胞在添加10%胎牛血清和抗生素的DMEM中培养。
胆管细胞增殖与死亡
将培养的胆管细胞(~50%合流)去除血清过夜,用1%胎牛血清、10%胎牛血清或TWEAK (20 ng/ml)刺激24小时。细胞计数。在BrdU存在的情况下,用FBS或TWEAK刺激胆管细胞24小时,并用抗BrdU抗体染色。将胆管细胞(~80%合流度)在不含FBS的DMEM或添加10% FBS和200 μM棕榈酸盐的DMEM中培养24 h。采用TUNEL或MTT法评估胆管细胞死亡和存活。
胆管细胞条件培养基和胆管细胞/BMDM共培养
融合性的尼克+/+和尼克−/−胆管细胞在添加1% FBS和50 ng/ml TWEAK的DMEM中培养8 h。用新鲜的DMEM (1% FBS,不含TWEAK)替换培养基,24 h后收集(胆管细胞条件培养基)。从C57BL/6J雄性小鼠的股骨和胫骨中分离BMDMs,在添加20% L929条件培养基、10%热灭活胎牛血清、100单位/ml青霉素和100µg/ml链霉素的DMEM中培养15.6天后bmdm完全分化。分化后的BMDMs在胆管细胞条件培养基中培养6 h,采用qPCR检测其基因表达。造血干细胞在添加5%牛血清、100单位/ml青霉素和100µg/ml链霉素的DMEM中培养。融合造血干细胞在添加50 ng/ml tgf - β1的胆管细胞条件培养基中培养24 h,采用qPCR检测其基因表达。在transwell中,将胆管细胞接种于插入物中,用添加1% FBS和50 ng/ml TWEAK的DMEM预处理8 h,然后在底孔中与添加20% L929条件培养基、10%热灭活FBS、100单位/ml青霉素和100µg/ml链霉素的DMEM中培养的BMDMs共培养。与胆管细胞共培养24 h后收获BMDMs,采用qPCR检测其基因表达。
胆管细胞培养中NIK的稳定性
用β-半乳糖苷酶(β-gal)或NIK腺病毒载体转导胆管细胞2小时,然后在添加5%胎牛血清的DMEM中培养16小时。用添加DDC (100 uM)、ANIT (50 uM)或DMSO的DMEM代替培养基,4 h后,用环己亚胺(100 ug/ml)处理细胞0.5、1、1.5、2、2.5和3 h。制备细胞提取物,并用NIK和β-肌动蛋白抗体进行免疫印迹。NIK水平被量化,归一化为β-肌动蛋白,并以初始值的百分比表示。
实时定量PCR
总rna采用TRIzol试剂(Life technologies)提取。使用Radiant™SYBR Green 2X Lo-ROX qPCR试剂盒(Alkali Scientific, Pompano Beach, FL)和StepOnePlus RT PCR系统(Life Technologies Corporation, NY, USA)测量相对mRNA丰度。引物列于补充表2.
统计数据
数据采用Microsoft excel 2016进行分析,以均数±sem表示。采用双尾Student’s分析两组间的差异t纵向资料采用双因素方差分析/Sidak后验检验(GraphPad Prism 8)。P< 0.05认为有统计学意义。
报告总结
有关研究设计的进一步资料,请参阅自然研究报告摘要链接到这篇文章。
数据可用性
源数据在本文中作为源数据文件提供。支持本文所述发现的所有数据均可在文章和补充信息中获得,并可根据合理要求从通讯作者处获得。源数据都提供了这篇论文。
参考文献
Cardinale, V.等。人胆道树中的多能干细胞/祖细胞产生肝细胞、胆管细胞和胰岛。肝脏病学54, 2159-2172(2011)。
Gouw, A. S, Clouston, A. D.和Theise, N. D.人类肝脏的导管反应:界面的多样性。肝脏病学54, 1853-1863(2011)。
巴纳莱斯,j.m.等。Cholangiocyte病理学。Nat Rev. Gastroenterol。乙醇。16中文信息学报,269-281(2019)。
Sancho-Bru, P.等。肝祖细胞标记物与酒精性肝炎患者肝损伤相关并预测短期死亡率肝脏病学55, 1931-1941(2012)。
Bonkovsky, H. L.等。药物、草药和膳食补充剂引起胆管损失的临床表现和结果。肝脏病学65, 1267-1277(2017)。
Vedeld, h.m.等人。胆汁甲基化标志物对原发性硬化性胆管炎患者胆管癌的早期准确检测。肝脏病学75, 59-73(2022)。
白,H.等。yes相关蛋白调控胆管结扎后肝脏反应。肝脏病学56, 1097-1107(2012)。
Santamaria, E.等。表皮生长因子受体配体双调节蛋白保护胆汁淤积性肝损伤并调节胆汁酸合成。肝脏病学69, 1632-1647(2019)。
菲克特,P.等。一种新的异种生物诱导的硬化性胆管炎和胆道纤维化小鼠模型。点。j .分册。171, 525-536(2007)。
非典型NF-kappaB通路。Immunol。牧师。246, 125-140(2012)。
Jakubowski等人。TWEAK诱导肝祖细胞增殖。j .中国。投资.115, 2330-2340(2005)。
伯德,t.g.等。在没有损伤的情况下,骨髓注射通过巨噬细胞介导的TWEAK信号通路刺激肝小管反应。自然科学进展。美国110, 6542-6547(2013)。
Tirnitz-Parker, j.e.等。肿瘤坏死因子样弱凋亡诱导剂是肝祖细胞的有丝分裂原。肝脏病学52中文信息学报,291-302(2010)。
盛磊,等。nf - kappab诱导激酶(NIK)通过增强胰高血糖素的作用促进肥胖的高血糖和葡萄糖耐受不良。地中海Nat。.18, 943-949(2012)。
Shen, H.等。小鼠肝细胞过度表达nf - kappab诱导激酶(NIK)可引发致命的巨噬细胞依赖性肝损伤和纤维化。肝脏病学60, 2065-2076(2014)。
Ling, L, Cao, z, & Goeddel, D. V. nf - kappab诱导激酶通过磷酸化Ser-176激活ikk - α。自然科学进展。美国95, 3792-3797(1998)。
Malinin, N. L., Boldin, M. P., Kovalenko, A. V.和Wallach, D. map3k相关激酶参与TNF, CD95和IL-1诱导NF-kappaB。自然385, 540-544(1997)。
Senftleben, U.等。IKKalpha激活第二种进化保守的NF-kappa B信号通路。科学293, 1495-1499(2001)。
肖国强,孙淑娟,孙淑娟,孙淑娟。nf - kappab诱导激酶调控nf - kappab2p100的表达。摩尔。细胞7[j], 2001。
Jijon, H., Allard, B. & Jobin, C. NF-kappaB诱导激酶通过p38 mapk依赖的RelA磷酸化途径独立于IkappaB激酶γ激活NF-kappaB转录活性。手机信号16, 1023-1032(2004)。
熊艳,等。肝nf - kb诱导激酶(NIK)抑制急性和慢性肝病小鼠肝脏再生。Elife7, e34152(2018)。
nf - κ b诱导激酶:免疫系统和癌症的关键调节因子。细胞因子生长因子21, 213-226(2010)。
Shen, H.等。胸腺nf - kappab诱导激酶(NIK)调节小鼠CD4+ T细胞引起的肝损伤和纤维化。J乙醇。67, 100-9(2017)。
Shen, H.等。骨髓胸腺上皮nf - kb诱导激酶(NIK)/IKKalpha通路影响小鼠自身免疫和肝、肺稳态。自然科学进展。美国116, 19090-19097(2019)。
钟旭,等。肝NF-kappaB诱导激酶和NF-kappaB激酶亚单位α抑制剂可促进肝脏氧化应激、铁下垂和肝损伤。乙醇。Commun。5, 1704-1720(2021)。
赵安、雷、康春、顾国强、许玉娟、陈志强、陈志强等。一种CK19(CreERT)敲入小鼠细胞系可在多种内胚层器官上皮细胞中进行条件性DNA重组。《创世纪》46, 318-323(2008)。
Liu, Y.等。肝脏nf - kappab诱导激酶(NIK)促进肥胖雄性小鼠肝脏脂肪变性和葡萄糖反调节。内分泌学1581207 - 16(2017)。
Natarajan, s.k.等。饱和游离脂肪酸诱导胆管细胞脂肪凋亡。肝脏病学60, 1942-1956(2014)。
Castanedo, g.m.等人。基于结构设计的三环NF-kappaB诱导激酶(NIK)抑制剂对磷酸肌苷-3激酶(PI3K)具有高选择性。医学化学。60, 627-640(2017)。
周毅,等。brahma相关基因1抑制通过减弱祖细胞扩张来预防肝纤维化和胆管癌。肝脏病学74, 797-815(2021)。
埃斯纳,C.等。RELB的核易位在患病的人肝脏中增加,并促进小鼠的管状反应和胆道纤维化。胃肠病学156, 1190 - 1205。e1114(2019)。
Syal, G., Fausther, M.和Dranoff, J. A.胆管细胞免疫生物学进展。点。j .杂志。Gastrointest。肝脏杂志。303, g1077-g1086(2012)。
Liu, B.等。IKKalpha是维持皮肤稳态和预防皮肤癌所必需的。癌症细胞14中文信息学报,212-225(2008)。
致谢
我们感谢dr。Yatrik Shah, Xin Tong和Lei Yin(密歇根大学)进行了有益的讨论。本研究由美国国立卫生研究院R01 DK114220、R01 DK115646、R01 DK127568、R01 DK130111和R21 AA025945 (L.R.)、R01 DK116548 (M.B.O.)、UH3 AA026903 (S.L.)和密歇根大学GI创新基金资助。这项工作利用了密歇根糖尿病研究和培训中心(NIH DK020572)、密歇根代谢组学和肥胖中心(DK089503)和密歇根大学肠道肽研究中心(NIH DK34933)支持的核心。
作者信息
作者及单位
贡献
本实验由郑正正、张正正、张正正、张正正、张正正、张正正设计实验并撰写论文,张正正、张正正、张正正、张正正、张正正、张正正、张正正、张正正、张正正、张正正、张正正、张正正和张正正编辑论文,张正正、张正正、张正正和张正正提供人体肝脏活检。
相应的作者
道德声明
相互竞争的利益
作者声明没有利益冲突。
同行评审
同行评议信息
自然通讯感谢匿名审稿人对这项工作的同行评审所做的贡献。同行评审报告是可用的。
额外的信息
出版商的注意b施普林格《自然》杂志对已出版的地图和机构的管辖权要求保持中立。
源数据
权利和权限
开放获取本文遵循知识共享署名4.0国际许可协议,该协议允许以任何媒介或格式使用、共享、改编、分发和复制,只要您适当地注明原作者和来源,提供知识共享许可协议的链接,并注明是否进行了更改。本文中的图像或其他第三方材料包含在文章的知识共享许可协议中,除非在材料的署名中另有说明。如果材料未包含在文章的知识共享许可中,并且您的预期用途不被法律法规允许或超过允许的用途,您将需要直接从版权所有者处获得许可。要查看此许可证的副本,请访问http://creativecommons.org/licenses/by/4.0/.
关于本文

引用本文
张忠,钟翔,沈浩。et al。胆道NIK促进小管反应和小鼠肝损伤及纤维化。Nat Commun13, 5111(2022)。https://doi.org/10.1038/s41467-022-32575-8
收到了:
接受:
发表:
DOI:https://doi.org/10.1038/s41467-022-32575-8
