








摘要
上新世晚期和更新世早期,距今360万至80万年前1气候是否与未来变暖预测的气候相似2.古气候记录显示强烈的极性放大,年平均温度比当代值高出11-19°C3.,4.在此期间居住在北极的生物群落仍然鲜为人知,因为化石很少5.在这里,我们报告了一个古老的环境DNA6(eDNA)记录描述了格陵兰岛北部Kap København组丰富的植物和动物组合,可追溯到大约200万年前。该记录显示了一个开放的北方森林生态系统,其中有杨树、桦树和山茱萸的混合植被,以及各种北极和北方灌木和草本植物,其中许多是以前没有在大化石和花粉记录中发现的。DNA记录证实了兔子和线粒体DNA的存在,这些动物包括乳齿象、驯鹿、啮齿动物和鹅,都是它们今天和晚更新世亲戚的祖先。包括马蹄蟹和绿藻在内的海洋物种的存在支持了比今天更温暖的气候。重建后的生态系统没有现代的类似物。这种古老的eDNA的存活可能与它与矿物表面的结合有关。我们的发现开辟了基因研究的新领域,证明了使用古代eDNA追踪200万年前生物群落的生态和进化是可能的。
主要
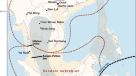
Kap København地层位于格陵兰岛北部(北纬82°24′、西经22°12′)的Peary Land,现在是一个极地沙漠。上层沉积层序包含保存完好的陆生动物和植物遗骸,在较温暖的早更新世间冰期循环期间被冲进河口7(无花果。1).该遗址近40年的古环境和气候研究提供了一个独特的视角,了解该遗址位于北极北部交错带的时期,重建的夏季和冬季平均最低温度分别为10°C和- 17°C,比现在高出10°C以上7,8,9,10,11.这些条件一定促使格陵兰冰盖大量消融,可能产生了最后的无冰间歇期之一7在过去240万年(迈)虽然已知Kap København组有保存完好的针叶林大化石和丰富的昆虫动物群,但很少发现脊椎动物的痕迹。迄今为止,这些包括遗存的lagomorph属,他们的粪和Aphodius甲虫,生活在哺乳动物的粪便中10,11.然而,加拿大北极地区埃尔斯米尔岛约3.4亿年前的Fyles Leaf床和海狸池保存着哺乳动物的化石,这些哺乳动物可能曾在格陵兰岛定居,例如已经灭绝的熊(Protarctos abstrusus)、已灭绝的海狸(DipoidesSp .),小犬科动物Eucyon以及北极巨型骆驼线4,12,13(类似于Paracamelus).纳勒斯海峡是否足以成为将格陵兰岛北部与这些动物隔离开来的屏障,仍然是一个悬而未决的问题。

Kap København组被正式划分为两个部分7(无花果。1).较低的A段由长达50米的层状泥浆组成,在近海冰川海洋环境中沉积有北极介形虫、有孔虫和软体动物14.上覆层段B由40-50米的砂质(单元B1和B3)和粉质(单元B2)沉积物组成(扩展数据图)。1),包括薄层,有机质丰富,间冰期大型动物化石沉积,沉积于靠近海岸的浅海或河口环境,以上、下滨面沉积相为代表7.
具体的沉积环境也反映在单元的矿物学中,其中近端的B3产地粘土含量最低,石英含量最高(样品组成见补充表)4.2.1而且4.2.2和补充表中的单位平均数4.2.3而且4.2.4).盆地充填物的结构表明,B组单元向现今海岸方向增厚,即向北部低山沉积物源的远端增厚。1).B1和B3单元记录了丰富的有机碎屑层,其中还含有丰富的北极和北方植物、无脊椎大化石以及陆生苔藓10,15.因此,DNA的埋藏学很可能反映了从一系列栖息地侵蚀而来的生物群落,它们被河流运输到前海岸,并在B1和B3单元内以有机碎屑混入沙质近岸沉积物的形式集中。相反,A段和B2单元的水相越深,海洋信号越强。Kap København组沉积物和Kim Fjelde组沉积物在矿物组成上的相似性支持了这一假设(补充表)4.2.1而且4.2.5).
地质时代
一系列的补充研究已经将Kap København组的沉积年龄范围从4.0-0.7 Myr缩小到2.4 Myr左右的2万年1- - - - - -3.).这是通过结合古地磁、生物地层学和同体地层学来实现的7,14,16,17,18.值得注意的是,地层记录中哺乳动物、有孔虫和软体动物的最后出现数据显示其年龄接近2.4 Myr(见补充资料,第4节)2).在这一总体框架内,我们添加了新的古地磁数据,显示A段磁极反转,而上覆单元B2的主要部分磁极正常。在之前的工作中,这与早更新世存在反转的三个磁地层层段是一致的:1.93 Myr(情景1),2.14 Myr(情景2)或2.58 Myr(情景3)(补充信息,第3节)1).此外,我们用宇宙成因来限制年龄26艾尔:10在本研究的四个地点确定成员B的埋葬年代(补充资料,第3.).Kap København组推荐的最大埋藏年龄为2.70±0.46 Myr。2;方法)。然而,我们抛弃了旧的设想3,因为它与在单一冰期-间冰期沉积旋回期间横跨a和B的持续沉积的证据相矛盾7,14,16,18,19.这就剩下了两种可能的情况(情况1和2),其中情况1支持年龄为1.9千万日,情况2支持年龄为2.1千万日。

一个修订后的古地磁分析显示B2单元具有正常的极性,并解锁了三种可能的年龄场景(S1-S3),包括成员A(蓝色)和B(棕色)。正常极性用黑色表示,反向极性用白色表示。是的,Jaramillo;科布山;Ol,奥杜威;菲,芬尼酒;Ka, Kaena;妈,猛犸。b、海洋有孔虫的存在和最后出现基准Cibicides grossus, rabbit-genusHypolagus还有软体动物北极蛤属多毛分别在北极、北半球和北格陵兰岛。最右边的蓝色带是根据贝壳上的氨基酸比例估计出的A成员的年龄范围7.c,两种不同生产比(7.42(黑色)和6.75(蓝色))计算的宇宙成因埋藏年龄的卷积概率分布函数。虚线和实线分别表示稳定侵蚀和零侵蚀的分布。这些分布都是最大年龄。d,分子测年桦木属sp.得出沉积物中DNA的中位年龄为1.323 Myr,其须限制95%高度后验密度(HPD)为0.68至2.02 Myr(蓝色密度图),运行马尔科夫链蒙特卡罗估计进行1亿次迭代。红点是利用乳齿象线粒体基因组限制放射性碳定年标本发现的中位分子年龄估计,而绿色区域包括BEAST中的分子钟估计标本,运行马尔科夫链蒙特卡罗估计4亿次迭代。胡须限制95%的火奴鲁鲁pd。
DNA保存
由于微生物的酶活性、机械剪切和自发的化学反应(如水解和氧化),DNA随时间而降解20..迄今为止所获得的最古老的DNA是从一颗永久冻土保存的猛犸象臼齿中恢复的,使用地质方法测定的年代为1.2-1.1 Myr,使用分子钟测定的年代为1.7 Myr(95%最高后密度,2.1-1.3 Myr)21.为了探索从Kap København组沉积物中回收DNA的可能性,我们计算了Kap København组DNA的热年龄及其预期的脱嘌呤程度。使用平均温度22(MAT)在−17°C时,我们发现DNA在恒定10°C下的热年龄为2.7千年,比2.0 Myr的年龄小741倍(补充信息,部分)4及补充表4.1.1).利用恐鸟化石的脱嘌呤率23,我们发现平均大小为50个碱基对(bp)的DNA可能在Kap København组中存活,假设该遗址保持冻结状态(补充信息,节)4及补充表10/24/11).在沉积物中保存DNA的机制可能不同于骨骼。矿物表面的吸附改变了DNA构象,可能阻碍了酶的分子识别,这有效地阻碍了酶降解24,25,26,27.为了研究在Kap København组中发现的矿物是否在沉积过程中保留并保存了DNA,我们使用x射线衍射确定了沉积物的矿物组成,并测量了它们的吸附能力。我们的研究结果强调,海洋沉积环境有利于细胞外DNA在矿物表面的吸附(补充信息,第2节)4及补充表4.3.1.1).与非粘土矿物(59-75 wt%)相比,粘土矿物(9.6-5.5 wt%)和蒙脱石(1.2-3.7 wt%)具有更高的吸附能力。DNA浓度代表了自然环境28(4.9 ng ml−1DNA),蒙脱石对DNA的吸附能力是石英的200倍。我们采用了沉积eDNA提取方案29从蒙脱石中只提取了5%的吸附DNA,从其他粘土矿物中提取了约10%(方法和补充信息,章节4).相比之下,我们提取了大约40%吸附在石英上的DNA。不同矿物的吸附能力和萃取率的差异表明矿物成分可能在古eDNA的保存和提取中起重要作用。
Kap København宏基因组
我们提取DNA29来自Kap København组5个不同地点的41个富含有机物的沉积物样本(补充信息,section)6来源数据1),转化为65个双索引Illumina测序文库30..首先,我们对65个植物质体DNA文库中的34个进行了筛选,筛选出保守的光系统II D2 (psbD使用ddPCR(液滴数字PCR)技术对基因进行检测,该基因靶向引物和探针跨越39 bp区域,并使用P7指数引物。此外,我们筛选了psbA基因使用类似的试验针对Poaceae(方法和补充图。6.12.1).在测试的34个样本中,有31个样本的清晰信号证实了这些文库中存在植物质体DNA(来源数据1,表5和6)。此外,我们使用Arctic古芯片1.0对65个文库中的34个进行了哺乳动物mtDNA捕获富集31使用Illumina HiSeq 4000和NovaSeq 6000对所有文库(初始和捕获)进行了鸟枪测序。共测序16,882,114,068个reads,经过适配器修剪、过滤≥30 bp、phred质量最小为30以及重复去除后,共测序2,873,998,429个reads。这些都是为了k -使用simka进行Mer比较32(补充资料,章节6),然后利用HOLI的竞争映射进行分类(https://github.com/miwipe/KapCopenhagen.git),其中包括最近发表的超过1500个北极和北方植物分类单元的基因组概要数据集33,34(方法和补充信息,章节6).考虑到样本的年龄以及与最近参考基因组的潜在遗传距离,我们允许每个read的相似性在95-100%之间,以便使用ngsLCA对其进行分类35.元admg (v.0.14.0)程序36随后用于量化和筛选所有宏基因组样本的死后DNA损伤的每个分类节点(方法)。该方法估计了终端位置的平均损伤(D-max)和似然比(λ-LR),它量化了损伤模型(即在读取开始时的更多损伤)与空模型(即常量损伤;参见“补充信息”部分6).我们发现DNA损伤显著增加,特别是对于真核生物(平均D-max = 40.7%,参见补充信息,第6节)。由此,我们将D-max≥25%作为分类节点的过滤阈值,用于进一步的下游分析以及λ-LR大于或等于1.5。此外,我们还设置了一个阈值,要求每个分类单元的最小读取数超过所有分类单元分配的读取数的中位数(除以2),以筛选低丰度分类单元。类似地,对于要考虑的样本,样本的总读取数必须超过每个样本读取数的中位数除以2,以过滤读取数最少的样本。最后,我们过滤出重复次数少于3次的分类群,随后通过转换成比例对读取结果进行归一化(图2)。3.而且4).

宏基因组中发现的植物组合的分类学剖面图。加粗的分类单元是仅以DNA形式存在的属,而不是大化石或花粉。星号表示在其他上新世北极遗址发现的。通过大化石或系统发育位置确定的灭绝物种用匕首标记。阅读分类为Pyrus而且马吕斯用磅符号标记,并且可能是属于蔷薇科中另一个物种的过度分类的DNA序列,不作为参考基因组存在。

一个, B1、B2和B3单元动物组合的分类概况。粗体的分类单元是仅在DNA中发现的属。b,系统发育安置和pathPhynder62线粒体读数的结果被唯一地分类为象皮科或象皮科以下(来源数据1)。通过大化石或系统发育位置识别的灭绝物种用匕首标记。
DNA,花粉和大化石的比较
格陵兰岛的海岸从北纬60°延伸到83°,包括从亚北极到北极沙漠的生物气候带37,38.格陵兰有175种原生维管植物属,不包括历史上引进的物种39,40,41.其中70例(40%)通过宏基因组分析检测到。3.);这些属中的大多数今天局限于Kap København的极地沙漠以南的生物气候带(见参考文献)。42以及其中的参考文献),例如,所有水生大型植物。指定阅读柳树,新,Vaccinium,桦木属,苔属植物而且木贼属在这些属中,木贼属,新,柳属北极蛤属还有两种苔属植物(苔属植物nardina而且苔属植物施坦斯)目前在那里生长,而只有少数的记录Vaccinium uliginosum在80ºN以上,和桦木属娜娜在74ºN以上(参考。43).在Kap København古eDNA组合中检测到的102个属中,39%不再生长在格陵兰岛,但在北美北方林木中存在(例如,云杉而且杨树)和北部落叶林和海洋林(例如,Crataegus,水松,金钟柏而且Filipendula).这种多样化组合中的许多植物属都不生长在永久冻土基底上,它们所需要的温度比今天格陵兰岛任何纬度的温度都高。
除DNA外,我们还对B3单元119号地点的6个样品进行了花粉计数。5.1.1).其中4个样品的花粉量为71 ~ 225粒(平均170.25)。样本4中约40%为山地草本植物,包括莎草科、ericaceae和蔷薇科。样本5和6以乔木类群为主桦木属.水螅类(例如,木贼属,Asplenium而且Athyrium filix-mas)和石松(石松属植物annotinum而且卷柏rupestris)也有很好的代表性,在样本1、4和6中占组合的30%以上。
在DNA鉴定的102个植物属中,共有39个属也在属水平上作为大化石或花粉出现。另有39个类群可能被鉴定为大化石或花粉,但不在同一分类学水平10,15(资料来源1,表1和2)。例如,Poaceae的12属经DNA鉴定(等,Anthoxanthum,Arctagrostis,Arctophila,Calamagrostis,Cinna,Dupontia,Hordelymus,Leymus,美丽姆,Phippsia而且《行动纲领》),只提供有关的资料Hordelymus在今天的北极已经找不到(http://panarcticflora.org/),但在花粉分析中仅在科水平上进行区分,仅发现一种禾本科大化石。有24个类群仅被记录为DNA。其中包括北方针叶树杨树还有少量灌木和矮灌木,但以草本植物为主。在73个植物属中发现了大型化石10,15在美国,只有24例在DNA分析中未被检测到。因为大化石和DNA有相似的埋藏体——都是在当地沉积的——它们之间比DNA和花粉之间有更多的重叠,而花粉通常是区域分散的44.DNA中缺失的9个类群是苔藓植物,可能是由于该类群在基因组参考数据库中代表性较差。此外,已灭绝的天南星科分类单元在参考数据库中不存在。其余未被发现的属为维管植物,除两个(胚胎学而且山茱萸)在大型化石记录中是罕见的。因为在大化石和DNA记录中,稀有类群的探测都具有挑战性45我们认为,基于这两种方法的局限性,DNA和大化石记录之间的这种重叠是可以预期的。
此外,在本尼克花粉记录中还记录了19个类群46包括四种乔木或灌木,五种蕨类,三种藓类,藻类,真菌和苔类各一种。我们还发现了来自风媒植物的花粉,尤其是裸子植物,它们可以分布在植物实际生长地区的极北地区10.Bennike46还注意到高比例的藓类和蕨类,并建议他们可能过度代表由于他们的孢子壁是抵抗退化。此外,如果这些类群优先分布在流入河口的河流中,它们的孢子在冲积层中的浓度可能比分布更广泛的类群的花粉更集中。因此,抗衰变性和冲积沉积都可能有助于我们观察到的相对频率。同样的冲积动力也可能导致了非常大的读取计数柳树,桦木属,杨树,苔属植物而且木贼属在宏基因组记录中,这意味着无论是这些类群在花粉记录中的比例,还是reads计数,都不一定与它们在区域植被中的实际丰度(生物量或盖度)相关。
最后,我们试图通过叶绿体DNA的系统发育位置来确定植物DNA的年龄。我们检查了该属的数据桦木属,杨树而且柳树因为它们具有足够高的叶绿体基因组覆盖率(平均深度分别为24.16×、57.06×和27.04×)和足够多的现代全叶绿体参考序列(方法)。由于它们的年龄和与现代参考基因组的潜在遗传距离,我们将唯一分类reads的相似度阈值降低到90%,并按单位合并以增加覆盖率。这两个桦木属而且柳树基本放置到大多数代表物种在各自属,和杨树放置结果显示支持不同物种的混合有关p . trichocarpa而且p . balsamifera(扩展数据图。7- - - - - -9).
我们使用桦木属叶绿体可用于分子年代测定分析,因为它们被确定地放置在系统发生树的单一边缘上(也就是说,不像在杨树),具有大量的参考序列,在古样本中具有较高的覆盖率。我们使用了BEAST47V1.10.4获取分子钟的日期估计为我国古代桦木属叶绿体样本(详见方法,“分子测年方法”)。我们包括了31个现代的桦木属和一个赤杨皮叶绿体参考序列,仅使用在古代样本中深度至少为20的位点,并包括先前估计的Betula-Alnus叶绿体分化时间4861.1 Myr用于根节点的校准。我们的BEAST分析对古代样本年龄的不同先验和不同的核苷酸取代模型都是稳健的(扩展数据图。10).由此得出的年龄中位数估计为1.323 Myr, 95% HPD为(0.6786,2.0172)Myr(图。2).
动物DNA结果
后生动物线粒体和核DNA记录的多样性远低于植物,但包含一个已灭绝的科,一个在今天的格陵兰岛已经没有了,四个脊椎动物属原产于格陵兰岛,以及四个无脊椎动物科的代表。4).赋值基于参考基因组的不完整和可变表示,因此我们将reads确定为科级,并且只有在存在足够线粒体reads的情况下,我们将这些reads匹配到基于更完整的现代线粒体序列的线粒体系统发育,从而将赋值细化到属级(补充信息,第2节)6).对于植物reads,相似性超过90%的唯一分类动物reads按单位进行解析和合并,以增加系统发育放置的覆盖率。
最值得注意的是,我们在单元B2和B3中发现了属于象科的读数,包括大象和猛犸象,但在分类学上不包括乳齿象(Mammutsp.)——然而,在NCBI分类法中,因此我们的分析读起来归类为象皮科或以下,因此包括Mammut我们象皮科线粒体基因组的共识读数落在Mammut分支(图;4 b),生长在乳齿象所有分支的基部。然而,我们注意到乳齿象中的这种位置仅依赖于两个过渡单核苷酸多态性(SNPs),第一个有三个读取深度,第二个只有一个(扩展数据图)。4方法和补充信息,章节6).此外,我们尝试用BEAST测定恢复的乳齿象线粒体基因组的年代49.我们采用了两种测年方法,一种是单独使用放射性碳测定标本,另一种是使用放射性碳和分子测定乳齿象。第一种分析得出了我们乳齿象有丝分裂基因组的中位年龄估计值为1.2 Myr (95% HPD: 191,000 yr-3.27 Myr),第二种方法得出的中位年龄估计值为5.2 Myr (95% HPD: 1.64-10.1 Myr)(补充图。6.8.5及补充资料部分6).
同样地,分配给子宫颈科的读数支持在基底上的位置至今(驯鹿和北美驯鹿)分支(扩展数据图。3.).线粒体读取映射到Leporidae(野兔和家兔)的位置接近欧亚野兔分支的基础(扩展数据图)。2),是唯一在化石记录中发现的哺乳动物7.天兔座,特别是天兔座arcticus它也是现今生活在格陵兰岛的Leporidae中唯一的属。蟋蟀科的线粒体reads仅覆盖了一个信息性的SNP,这表明它们来自Arvicolinae亚科(田鼠、旅鼠和麝鼠)(扩展数据图)。6).对于我们的数据中所代表的唯一的鸟类分类单元——鹅科,鹅和天鹅科,我们发现了该属的一个强大的基础位置Branta由3个读深度在2到4之间的转位snp支持(扩展数据图。5).与植物相比,基于线粒体参照的精细脊椎动物分配在生物地理上更为保守。Dicrostonyx中的Dicrostonyx groenlandicus(新北极领旅鼠)-是今天格陵兰岛本土唯一的蟋蟀科属,就像至今中的tarandus groenlandicus(贫瘠土地上的驯鹿)是鹿科唯一的成员。乳齿象是个例外,因为象皮科的任何一种动物都没有生活在今天的格陵兰岛。
来自海洋生物的远古DNA
在DNA记录中确认的其他后生动物类群是一种造礁珊瑚(Merulinidae)和几种节肢动物,与两种昆虫——蚁科(蚂蚁)和普利科(跳蚤)——以及一个海洋科——鲎科(马蹄蟹)相匹配。这有点出乎意料,因为Kap København组有丰富的昆虫大化石记录,其中包括200多个物种,包括福米卡海洋分类单元比陆地分类单元数量少,在海洋后生动物中未发现线粒体DNA。读取长度、DNA损伤以及分配的读取在参考基因组中均匀分布的事实表明,这些不是人工制品,而是可能是密切相关的、潜在灭绝物种的过度匹配DNA序列,这些物种目前由于分类学表现不佳而从我们的参考数据库中缺席。相比之下,狐鳞科,在亚门螯肢动物门,不太可能被错误识别,因为这个独特的属是其目的中唯一幸存的成员,因此与其他现存的生物严重分化。
这些数据的可能来源是鲎波吕斐摩斯它是该属中唯一的大西洋成员,在沉积物积累时,它会直接在沉积物上繁殖。今天,该属不在芬迪湾(Bay of Fundy,约45°N)北部产卵,这表明Kap København早更新世较温暖的地表水条件与格陵兰岛东北部海岸更新世重建的+8°C年海面温度异常一致50.通过将读取值对准塔拉海洋真核生物宏基因组组装基因组(SMAGs)数据(方法),我们进一步揭示了在14个样本中存在24个海洋浮游类群,包括浮游动物和浮游植物(图2)。5).这些检测到的SMAGs属于超类群Opisthokonta(6)、Stramenopila(15)和Archaeplastida(3)。这些信号大部分来自现代海洋(即北冰洋和南大洋)寒冷地区的SMAGs,如硅藻(硅藻门)、Chrysophyceae和mast4类群(补充表)6.11.1),如我们所料。然而,一些是世界性的,而其他的,如古质体(绿色微藻),具有海洋信号,今天仅限于太平洋的温带水域(图2)。5).尽管我们不知道现代生态系统是否可以外推到古代生态系统,但据信,在北极地区,绿色微藻的丰度正在增加,这往往与地表水变暖有关。

讨论
Kap København古eDNA记录的不同寻常有几个原因;估计分子年龄的95%最高后验密度的上限为2.0 Myr,独立支持约2 Myr的地质年龄(图2)。2).这意味着该DNA比任何先前测序的DNA都要古老得多21.我们的DNA结果检测到的植物属数量是之前使用鸟枪测序古沉积物的研究的五倍29,34,51,52,这完全在最丰富的北方北方元条形码记录的范围内53.76%的属或科级别的分类单元也出现在同一单元的大化石和/或花粉组合中,这一观察结果加强了分配的准确性。我们的结果证明了古代环境宏基因组学在重建古代环境、系统发育位置和距今约2 Ma的不同类群的古代谱系方面的潜力(补充信息,节)6).最后,DNA鉴定了一组额外的植物属,它们作为大化石出现在其他北极地区的晚上新世和早更新世遗址(图2)。1而且3.及补充资料部分5),而不是Kap København的化石,从而扩大了这些古代植物群的时空分布。
值得注意的是,两者的检测至今(驯鹿和北美驯鹿)和Mammut(乳齿象)迫使人们根据该遗址相对贫瘠的动物群记录,对早期的古环境重建进行修正,在沉积期的大部分时间里,这意味着更高的生产力和栖息地多样性。因为所有通过DNA鉴定的脊椎动物类群都是食草动物,所以它们的表示可能是相对生物量的函数(见补充资料,节中关于埋葬学的讨论)6).在北方的环境中,驯鹿、鹅、野兔和啮齿动物都是丰富的,至少是季节性的。此外,大型食草动物(如驯鹿,特别是乳齿象)的粪便可能是沉积物的重要组成部分34.相比之下,食肉动物没有出现,这与它们的总生物量较小一致。这种动态也解释了植物读类相对于后生动物的优势,以及在某种程度上不同植物属的表现差异(补充资料,章节6).在化石普遍缺失的情况下,DNA可能被证明是重建早更新世脊椎动物生物地理的最有效工具。乳齿象的DNA肯定暗示了这种大型食草动物的存活种群,这将需要一个比早期主要基于植物大型化石的重建推断的更多产的北方栖息地7.在新斯科舍省中部的一处遗址中,大约7.5万年前的乳齿象粪便中含有莎草、香蒲、芦苇、苔藓甚至是叶生植物的大型化石,但主要是云杉针和桦树54.带有乳齿象DNA的Kap København单元产生了大型化石和来自桦木属以及更多的嗜热树木类群,包括金钟柏,水松,山茱萸而且荚莲属的植物这些地区都没有延伸到今天格陵兰岛的北极苔原或极地沙漠。这些类群在多个单元中同时出现,迫使对以前的温度估计以及永久冻土的存在进行修正。
没有一个单一的现代植物群落或栖息地包括Kap København的许多大化石和DNA样本中所代表的类群范围。群落组合代表了现代北方和北极类群的混合物,这在现代植被中没有类似物10,15.在某种程度上,这是意料之中的,因为这些属的现代成员的生态振幅已经被进化改变了55.此外,北极高光周期与较暖条件和较低大气CO的结合2浓度56使北格陵兰岛的早更新世气候与今天非常不同。陆地组合的混合特征也反映在海洋记录中,北极和更国际化的蛇孔鱼和Stramenopila的SMAGs与马蹄蟹、珊瑚和绿色微藻(古原藻)一起被发现,它们今天生活在更南纬的温暖水域。
大型食草动物,尤其是乳齿象,可能对间冰期针叶林环境产生了重大影响,甚至对高纬度地区的植被结构和组成提供了自上而下的营养控制。乳齿象的存在57,58再加上没有人为火灾,这在一些全新世的北方栖息地发挥了作用59,是重要的区别。另一个重要因素是,当间冰期开始时条件变得有利时,先锋物种能够分散到北格陵兰岛的避难地的邻近性和生物的丰富性。早更新世冰川作用持续时间较短,产生的冰盖面积较小,使得加拿大东北部物种相对丰富的针叶落叶林地得以殖民12,60.更新世后期更广泛的冰川作用使北格陵兰岛越来越孤立,后来的重新殖民来自越来越遥远和/或多样性越来越少的难民。
总之,我们展示了古代eDNA的力量,为我们对这个独特的、与北极物种混合的古代北方开放森林群落的了解增加了大量细节,这个群落的组成没有现代类似物,包括乳齿象和驯鹿等。类似详细的植物群和脊椎动物DNA记录可能在其他地方保存下来。如果这些化石被发现,将有助于我们了解北极地区温暖的早更新世时期的气候变化和生物相互作用。
方法
抽样
沉积物样本采集于2006年、2012年和2016年夏季格陵兰岛北部Kap København组(82°24′00″N 22°12′00″W)(见补充表)3.1.1).采样材料包括富含有机物的永久冻土和干燥的永久冻土。在取样之前,对型材进行清洗以暴露新鲜材料。此后,使用直径10厘米的金刚石头钻头或切割出~40 × 40 × 40厘米的块,从山坡上垂直收集样本。沉积物在野外和运输到哥本哈根实验室设施的过程中被冷冻。使用一次性手套和手术刀,并在每个样品之间更换,以避免交叉污染。在受控的实验室环境中,岩芯和块进一步次采样,只取沉积物岩芯的内部部分,在内芯和表面之间留出1.5-2 cm,这提供了大约6-10 g的子样品。随后,所有样品保存在- 22°C以下的温度下。
我们通过在3个地层单元B1、B2和B3取样和生物复制,对富含有机物的沉积物进行采样,横跨5个不同的地点,地点:50 (B3)、69 (B2)、74a (B1)、74b (B1)和119 (B3)。进一步在不同的子层(编号L0-L4,源数据1,表1)中对每个站点的每个单元的每个生物副本进行采样。
绝对年龄测定法
2014年,用加速器质谱法分析了在现代深度(流切阶地下3至21米)收集的8× 1 kg富含石英的砂岩样品中的Be和Al氧化物目标,并使用简单的埋藏测年方法将宇宙成因同位素浓度解释为最大年龄1(26艾尔:10被规范化10)。的26艾尔和10Be同位素是在沉积之前,宇宙射线与Kap København上方山区风化层和基岩表面暴露的石英相互作用产生的。我们假设26艾尔:10在这些逐渐被侵蚀的古地表的上面几米,铍在很长一段时间内都是均匀稳定的。一旦被溪流和山坡过程侵蚀,石英砂就沉积在砂质辫状流沉积物、三角洲分流系统或近岸环境中,并有效地屏蔽了埋在沉积物、间歇冰架或冰盖覆盖下(至少在间冰期)的宇宙射线核子,直到最终出现。简单的埋藏年代测定方法假设沙粒只经历过一次埋藏事件。如果多次掩埋事件被重新暴露的时期分开发生,那么开始26艾尔:10在最后一次埋藏事件之前,由于相对较快的衰变,将小于初始生产比(6.75到7.42,见下文讨论)26,因此计算出的埋葬年龄将是最大限制年龄。多次埋藏事件可能是由于源区的厚冰川冰的遮挡,或在最终沉积之前由集水区的沉积物储存造成的。这些屏蔽事件意味着26艾尔:10Be较低,因此,假设初始生产比计算出的埋葬年龄会高估最终的埋葬时间。我们还认为,一旦被埋藏,沙粒可能已经暴露在次级宇宙成因μ子中(它们的深度对于海底核子的产生来说太大了)。由于在这些冰川覆盖的近岸环境中的沉积速度相对较快,我们表明,即使是μ子的产生也可以忽略不计补充信息).然而,一旦海洋沉积物出现在海平面以上,由核生和mugenic产生的原位生产可能会改变26艾尔:10是。的26艾尔和10Be等时线图揭示了这一复杂的埋藏历史(补充资料,第3.)和两者的浓度-深度复合剖面26艾尔和10我们发现,最浅的样本可能是在一段时间内(~15,000年前)暴露出来的,这与该地区的冰川消退相一致(补充信息)。虽然我们将所有样品的单个简单埋藏年龄解释为Kap København组B段的最大沉积极限年龄,但我们建议在单一深度剖面中使用三个屏蔽最深的样品,以最小化沉积后生产的影响。然后我们计算这三个样本(KK06A, B和C)的卷积概率分布年龄。然而,这个计算取决于26艾尔:10取决于我们使用的生产比率(即在6.75和7.42之间),以及我们是否调整了集水区的侵蚀。因此,我们对最低和最高的生产比以及0到最大的可能侵蚀率重复卷积概率分布函数年龄,以获得在1处的最小和最大限制年龄范围σ(补充信息,章节3.).取负3和正3之间的中点σ在置信范围内,得到的最大埋藏年龄为2.70±0.46 Myr。这三个样本在等时线图上的位置也支持这个年龄,这表明真实年龄可能与这个最大限制年龄没有显著差异。
热的年龄
Kap København DNA的热降解程度与来自Krestovka猛犸象臼齿的DNA进行了比较。公布的DNA降解动力学参数64用于计算长期温度记录在给定区间内的相对速率差异,并量化与10°C参考温度的偏移量,从而估算每个样品在10°C下的热年龄(补充信息,第4).Kap København沉积物的年平均气温(MAT)来自Funder et al. (2001)6对于Krestovka猛犸象,MAT是根据Cerskij气象站的温度数据计算的。251230)北纬68.80°161.28°E,距国际研究所数据库32米(https://iri.columbia.edu/)(补充表格4.1.1).
我们没有对Kap København沉积物或Krestovka猛犸象的热年龄计算进行季节波动校正。我们为Kap København沉积物中的DNA提供了四种不同热情景的理论平均片段长度(补充表)10/24/11).使用环境递减率(6.49°C km)对高度的热年龄计算进行了修正−1).我们缩放了Hansen等人(2013)的长期温度模型。65通过一个足以解释末次冰盛期当地温度下降估计的比例因子,对当前垫层的局部估计进行了评估,然后使用127 kJ mol的活化能(Ea)估算了综合速率−1(ref。64).
矿物学组成
每个Kap København沉积物样品中的矿物质都是用x射线衍射识别的,它们的比例是用Rietveld细化量化的。样品均质是通过在McCrone Mill中用乙醇研磨~ 1g沉积物10分钟。样品在60°C下干燥,加入刚玉(CR-1, Baikowski)作为内标,最终浓度为20.0 wt%。衍射图使用Bruker D8 Advance (Θ -Θ geometry)和LynxEye探测器(开口2.71°)收集Kα1、2辐射(1.54 Å;40 kV, 40 mA)使用衍射光束上厚度为0.2 mm的镍滤光片和设置为3mm的束刀。我们从5-90°2θ扫描,步长为0.1°,步长为4秒,样品以20转/分旋转。发散缝的开口为0.3°,反散射缝的开口为3°。主索勒裂隙和次索勒裂隙开度为2.5°,探测窗开度为2.71°。对于Rietveld分析,我们使用BGMN软件的Profex接口66,67.利用基本参数射线追踪法确定了仪器参数和峰展宽68.黏土矿物鉴定的详细描述可在辅助资料中找到。
吸附
我们使用纯矿物或纯化矿物进行吸附研究。所使用的矿物及其净化方法见补充表4.2.6.矿物的纯度是用x射线衍射检查的,仪器参数和程序与上一节中列出的相同,即矿物成分。关于起源、提纯和杂质的说明可以在补充信息部分找到4.我们用人工海水69和鲑鱼精子DNA(低分子量,冻干粉,Sigma Aldrich)作为eDNA吸附的模型。将已知量的矿物粉末与海水混合,在超声波浴中超声15分钟。将DNA原液加入悬浮液中,使最终浓度在20 ~ 800 μg ml之间−1.悬浮液在旋转振动筛上平衡4小时。然后将样品离心,用紫外光谱法(生物光度计,Eppendorf)测定上清液中的DNA浓度,同时进行阳性和阴性对照。所有的测量都是一式三份的,我们对每种矿物质测得5到8个DNA浓度。我们利用Langmuir和Freundlich方程将模型与实验等温线拟合,并获得了在给定平衡浓度下矿物的吸附容量。
花粉
花粉样品提取采用俄罗斯科学院地质研究所采用的改良Grischuk方案,该方案利用焦磷酸钠和氢氟酸70.用moticba 400复合显微镜在400倍放大下扫描6个样品的载玻片,并使用Moticam 2300相机拍摄。花粉百分比计算为包括未鉴定颗粒在内的总孢粉的比例。6个样品中只有4个样品的花粉数≥50。在这些样本中,鉴定出的孢粉总数在225到71之间(平均为170.25;中位数= 192.5)。使用几个已发布的密钥进行标识71,72.花粉图最初使用Tilia版本1.5.12编译73但在这项研究中使用Psimpoll 4.10重新绘制74.
DNA复苏
为了进行回收率计算,我们用DNA饱和矿物表面。为此,我们使用了与测定吸附等温线相同的方法,只是增加了一步,以去除未被吸附但仅困在湿糊体间质孔中的DNA。这一步骤很重要,因为间质DNA会增加明显吸附的DNA数量,从而高估了回收率。为了去除吸附后捕获的DNA,我们将矿物质重新分散在海水中。湿膏在海水中再分散、超离心和上清去除的过程不超过2.5 min。第二次离心后,将湿膏冷冻直至提取。我们使用了与Kap København沉积物相同的提取方案。提取后,再次用紫外光谱法测定DNA浓度。
基因组
共提取41份样本进行DNA检测75并转换为65个双索引Illumina测序库(包括13个阴性提取和库控制)30..34个文库随后使用QX200 AutoDG液滴数字PCR系统(Bio-Rad)按照制造商的协议进行ddPCR。ddPCR检测包括P7指数引物(5’-AGCAGAAGACGGCATAC-3’)(900nM)、基因靶向引物(900nM)和基因靶向探针(250nM)。我们筛选了Viridiplantae psbD(引物:5 ' - tcataattggacgttgaac -3 ',探针:5 ' -(FAM) actcccatatgaaa (BHQ1)-3 ')和Poaceae psbA(引物:5 ' - ctcacaacttccctctagac -3 ',探针5 ' -(HEX)AGCTGCTGTTGAAGTTC(BHQ1)-3 ')。此外,65个文库中的34个使用靶向捕获富集,用于哺乳动物线粒体DNA,使用古芯片Arctic1.0诱饵集31所有文库随后在Illumina HiSeq 4000 80 bp PE或NovaSeq 6000 100 bp PE上测序。我们总共测序了16,882,114,068个reads,经过低复杂度过滤(Dust = 1),质量修剪(问≥25),对大于29 bp的reads进行重复去除和过滤(仅对NovaSeq数据进行配对读对),结果有2,873,998,429个reads被解析用于进一步的下游分析。我们接下来估计k所有样本之间的Mer相似性使用simka32(设置最大读取次数的启发式计数(-max-reads 0)和ak(-kmer-size 31)),并对得到的距离矩阵进行主成分分析(PCA)补充信息“DNA”)。我们以后通过HOLI解析所有的QC读取33用于分类学分配。为了提高分类学分配的分辨率和灵敏度,我们用最近发表的北极-北方植物数据库(PhyloNorway)和北极动物数据库补充了RefSeq(92不含细菌)和核苷酸数据库(NCBI)34并在NCBI SRA中检索了139个北方动物类群基因组(2020年3月),其中发现并添加了16个部分全基因组(源数据1,表4),并使用GTDB微生物数据库95版作为诱饵。随后使用samtools合并所有比对,并使用z-sort进行排序(v. 1)。然后使用新开发的metaDMG估计胞嘧啶去氨频率,首先为每个读取找到所有可能比对的最低共同祖先,然后计算每个分类级别的损伤模式36(补充信息,部分6).同时,我们计算了平均读取长度以及每个分类节点的读取数(补充信息,部分6).我们对所有分类级别的DNA损伤分析表明,所有分类级别的所有样品都存在最小滤波器,D-max≥25%,似然比(λ-LR)≥1.5。这确保了只有显示古代DNA特征的分类群被解析用于下游分析,结果在任何对照中都没有发现分类群(补充信息,第2节)6).
海洋真核宏基因组
我们试图识别海洋真核生物,首先使用Kraken 2将所有质量控制的reads分类标记为真核生物、古细菌、细菌或病毒76使用参数“置信度0.5—最小命中组3”,并结合额外的过滤步骤,只保留根到叶评分为>0.25的读取。对于最初的Kraken 2搜索,我们使用了一个由taxdb-integration工作流创建的粗略数据库(https://github.com/aMG-tk/taxdb-integration)涵盖生命的所有领域,并包括海洋浮游真核生物的基因组数据库63其中包含683个宏基因组组装基因组(mag)和30个单细胞基因组(sag)塔拉海洋77,遵循Delmont等人的命名惯例。63,我们称之为smag。标记为根、未分类、古生菌、细菌和病毒的Reads通过Kraken 2标记的第二个步骤进行了细化,该步骤使用了由taxdb集成工作流创建的包含古生菌、细菌和病毒的高分辨率数据库。我们使用与初始搜索相同的Kraken 2参数和过滤阈值。Kraken 2数据库都建立了针对研究读取长度优化的参数(- kmer-len 25 -minimizer-len 23 -minimizer-spaces 4)。
标记为eukaryota, root和unclassified的Reads随后用Bowtie2进行映射78对SMAGs。我们使用了Picard的markduplicate (https://github.com/broadinstitute/picard)来删除重复项,然后我们用filterBAM程序(https://github.com/aMG-tk/bam-filter).进一步利用metaDMG中的贝叶斯方法对过滤后的BAM文件进行死后损伤估计,筛选出D-max≥0.25且拟合质量(λ-LR)大于1.5。去除reads小于500、平均reads平均核苷酸标识(ANI)小于93%、覆盖宽度比和覆盖均匀度小于0.75的SMAGs。我们采用数据驱动的方法来选择平均ANI读数阈值,我们探索了映射读数的变化作为平均ANI读数从90%到100%的函数,并确定了曲线中的弯头点(补充图2)。6.11.1).我们用anvi 'o79使用Delmont等人推断的SMAGs系统基因组树作为参考,在手动模式下绘制映射和损伤结果。我们使用Delmont等人的海洋信号作为每个海洋和海洋中SMAGs当代分布的代理。5及补充资料部分6).
DNA、大化石和花粉的比较
为了在DNA、大化石和花粉的记录之间进行比较,分类法按照泛北极植物区系清单进行了协调43和NCBI。例如,自Bennike(1990)以来18,Potamogeton已经分裂为Potamogeton而且Stuckenia,Polygonym已被拆分为蓼属植物而且Bistorta,虎耳草属被分割为虎耳草属而且Micranthes,而其他的则被合并了,比如Melandrium与硅宾40.植物科已经改变了名称——例如,禾草科现在被称为禾草科,而玄参科已经被重新划分,不包括车前草科和车前草科80.将该类群划分为:第1类为DNA和大化石或花粉记录的全部相同属,第2类为DNA记录的大化石或花粉记录的属,包括科级分类的属,第3类为DNA记录的类群,第4类为大化石或花粉记录的类群(来源资料1)。
系统发育位置
我们试图在系统发育学上对具有最多reads的古代类群进行定位,并使用足够数量的参考序列来构建系统发育。这些分类单元包括植物区系属的叶绿体基因组柳树,杨树而且桦木属,以及象皮科、蟋蟀科、白兔科、羊绒亚科和鹅绒亚科的线粒体基因组。虽然叶绿体基因组的进化不如植物线粒体基因组的进化稳定,但它的进化速度更快,而且不重组,因此比植物线粒体更有可能包含更多可供我们分析的信息位点81.像线粒体基因组一样,叶绿体基因组也有很高的拷贝数,因此我们可以预期有大量的沉积reads映射到它。
对于这些分类群,我们从NCBI Genbank下载了一组具有代表性的全叶绿体或全线粒体基因组fasta序列82,包括一个来自最近分离的外群的代表性序列。为桦木属我们还从PhyloNorway数据库中收录了三个叶绿体基因组34,83.我们将fasta文件中的所有模糊碱基都改为n。我们使用MAFFT84并在NCBI MSAViewer中检查了多个序列的比对,以确认质量85.我们通过去除MegaX中的d-loop,对Leporidae, Cricetidae和Anserinae的高度可变控制区进行了质量不足的线粒体校准86.
野兽套房49使用默认参数,从参考序列的多序列比对(msa)中为每组分类群创建超系统发育树,这些序列在Figtree中从Nexus格式转换为Newick格式(https://github.com/rambaut/figtree).然后,我们将多个序列对齐从BioPython传递给Python模块AlignIO87为每一组分类单元创建一个参考共识序列。此外,我们使用了SNPSites88从每个msa创建一个vcf文件。由于SNPSites在丢失数据时输出的格式与下游分析所需的格式略有不同,因此我们使用自定义R脚本适当地修改vcf格式。我们还过滤了非双等位基因的snp。
从损坏过滤的ngsLCA输出中,我们提取了在这些各自的分类单元中唯一分类为引用序列或分配给分类组中的任何共同祖先的所有readid,并使用seqtk (https://github.com/lh3/seqtk).我们合并来自所有站点和层的读取,为每个分类单元创建一个单独的读取集。接下来,由于这些提取的读取数据被映射到一个参考数据库,其中包括来自每个分类单元的多个序列,因此输出文件不在同一个坐标系统上。为了避免这个问题并避免映射偏差,我们使用bwa将每个读集重新映射到上面为该分类单元生成的共识序列89与古代DNA参数(bwa aln -n 0.001)。我们将这些读取转换为bam文件,删除未映射的读取,并使用samtools筛选映射质量> 2590.这产生了103,042,39,306,91,272,182和129的读数柳树,杨树,桦木属、象皮科和毛茛科。
接下来我们使用pathPhynder62,一种系统发育定位算法,从参考面板中识别系统发育上的信息性标记,评估重叠这些标记的古样本中的snp,并遍历树,根据其派生的和祖先的snp在每个分支上放置古样本。我们使用纯转换滤波器来避免由于脱amination导致的错误,除了桦木属,柳树而且杨树其中我们没有使用过滤器,因为覆盖面足够高。最后,我们研究了每个分类单元集中的pathPhynder输出,以确定我们古代样本的系统发育位置(关于系统发育位置的讨论,请参阅补充信息)。
在上述分析的基础上,进一步研究了其在属内的系统发育位置Mammut或乳齿象。为了避免下游结果的参考偏差,我们首先从上述分析中使用的所有比较线粒体基因组构建了一个共识序列,并将ngsLCA中鉴定为Elephantidae的reads映射到共识序列。共识序列首先使用MAFFT对齐所有感兴趣的序列84并在genious v2020.0.5中采用多数决定原则(https://www.geneious.com).我们对我们的序列进行了三个系统发育位置分析:(1)与每个象皮科物种(包括海牛)的单个代表进行比较(儒艮dugon(2)与每个象科物种的单个代表进行比较;(3)与包括亚洲象在内的所有已发表的乳齿象线粒体基因组进行比较作为事。
对于这些分析,我们首先使用BEAST v1.10.4构建了一个新的参考树。47)并重复前面描述的pathPhynder步骤,除了pathPhynder树路径分析MammutSNPs基于转换和反转,而不是由于低覆盖率而仅限于反转。
Mammut americanum
我们使用象皮科线粒体参考序列,GTR+G,严格时钟,出生-死亡替代模型,确认了我们序列的系统发育位置,并运行MCMC链2000万次,每20,000步采样一次。采用示踪仪评估收敛性91v1.7.2和有效样本量(ESS) > 200。为了确定我们恢复的乳齿象有丝分裂基因组的大致年龄,我们使用BEAST进行了分子测年分析47v1.10.4。正如最近发表的一篇文章所证明的那样,我们在测定乳齿象丝裂基因组的年代时使用了两种不同的方法92.首先,我们通过与放射性碳测定标本数据集(n只有13)。其次,我们估计了我们的序列的年龄,包括分子(n= 22)及放射性碳年代测定(n= 13)样本,使用先前确定的分子日期92.我们使用了与Karpinski等人相同的BEAST参数。92用gamma分布(5%分位数:8.72 × 10)设置样本的年龄4,中位数:1.178 × 106;95%分位数:5.093 × 106;初始值:74,900;形状:1;规模:1700000)。简而言之,我们指定了一个GTR+G4的替代模型,一个严格的时钟,恒定的种群大小,并运行马尔科夫链蒙特卡洛链50,000,000次运行,每50,000步采样一次。再次使用示踪剂确定井眼的收敛性。
分子测年法
在本节中,我们描述了古代桦树的分子年代测定(桦木属)叶绿体基因组使用BEAST v1.10.4(参考。47).原则上,属桦木属,杨树而且柳树具有足够高的叶绿体基因组覆盖率(平均深度分别为24.16×, 57.06×和27.04×,尽管这种覆盖率在叶绿体基因组中高度不均匀)和足够多的参考序列来尝试对这些样本进行分子测年。值得注意的是,这是我们在这些系统发育树中包括一个最近分化的外群的原因之一,其分化时间估计为每个系统发育树。然而,我们的杨树样本显然包含了不同物种的混合物,从它在pathPhynder输出中不一致的位置可以看出。特别是,两者都有多个支持snp杨树balsamifera而且杨树trichocarpa,以及以上分支上的支持和冲突snp。此外,经检查,我们的柳树样本中含有惊人的高数量的私有snp,这与任何古代甚至现代都不一致,特别是考虑到分配到系统发育树边缘的snp数量导致其他snp柳树序列。我们不确定造成这种不一致的原因,但假设我们的柳树样品也是混合样品,包含多个柳树在不同时期从系统发生树上同一位置分支分化的种。这可以通过观察覆盖这些私有SNP位点的所有读取来支持,这些读取通常来自混合样本,在许多情况下,包含替代和参考等位基因的读取比例很高。或者,或潜在的联合并行,这可能是大量的核质体DNA序列(nupt)的结果柳树93.正因为如此,我们才继续桦木属.
首先,我们下载了27篇完整的参考文献桦木属从NCBI基因库中提取了一个叶绿体基因组序列和一个桤木叶绿体基因组序列作为外群,并补充了三个桦木属叶绿体序列来自PhyloNorway数据库中最近的一项研究29,共31个参考序列。由于叶绿体序列是圆形的,下载的序列可能并不总是处于相同的方向或相同的起始点,因此我们使用自定义代码(https://github.com/miwipe/KapCopenhagen),它使用锚定字符串将参考序列旋转到相同的方向,并从同一点启动它们。我们用Mafft创建了这些转换后的参考序列的MSA84在Seqotron用眼睛检查了我们的对准质量94和NCBI MsaViewer。接下来,我们使用BioAlign共识函数从这个MSA调用一个共识序列87在Python中,它是多数原则的共识调用者。我们将使用这个一致序列来绘制古代桦木属读书要,既要避免参考偏颇,又要得到古今桦木属样品在与参考MSA相同的坐标上。
来自metaDMG中最后的公共祖先输出36,我们提取了所有单元、站点和级别的阅读集,这些单元、站点和级别被唯一分类到的分类级别桦木属或更低,最小序列相似度为90%或更高桦木属序列,使用Seqtk95.我们将这些读数集与共识相对应桦木属利用BWA进行叶绿体基因组分析89使用古DNA参数(-o 2 -n 0.001 -t 20),然后去除未映射的读取,质量过滤读取质量≥25,并使用samtools对得到的bam文件进行排序89.出于分子年代测定的目的,将这些读取集视为单个样本是合适的,因此我们使用samtools将得到的bam文件合并为一个样本。我们使用了bcftools89要制作一个mpileup并调用一个VCF文件,使用单倍体选项并禁用默认调用算法,这可以略微偏向于引用序列的调用,有利于通过默认的基本质量截断值13的基数上的大多数调用。我们包含了使用基本对齐质量的默认选项96,我们发现它大大降低了一些碱基的读深,并去除了indel区域周围的伪snp。最后,我们过滤了vcf文件,只包括单核苷酸变异,因为我们不相信其他变异,如在这种类型的古代环境样本中的插入或删除,有足够高的置信度来包括在分子测年中。
我们下载了最长的gff3注释文件桦木属参考序列MG386368.1,来自NCBI。使用自定义R代码97,我们解析了该文件和相关的fasta,将各个位点标记为蛋白质编码区域(其中我们根据gff3文件中记录的相位和链将碱基标记为其在密码子中的位置),RNA,或既不是编码也不是RNA。我们提取了编码区域,并输入了速控94在参考序列和古序列的相关位置上,他们翻译成一个很好的蛋白质比对(例如,没有过早的终止密码子)。尽管现代参考序列的编码区域被翻译成高质量的蛋白质比对,但在没有深度截断的古代序列中翻译相关位置会导致过早的终止密码子和整体低质量的蛋白质比对。另一方面,当使用深度截断20并将古序列中不符合该过滤器的位点替换为N时,我们看到了高质量的蛋白质比对(N位点除外)。我们还询问了古代序列中与共识不同的任何位置,并发现任何可疑区域(例如,基因组中多个snp在空间上紧密聚集在一起)都被深度截断20删除了。正因为如此,我们只在古代和现代样本中满足至少20个古代样本深度截断的遗址中进行了研究,这些遗址约占总数的30%。
接下来,我们通过多个序列对齐解析这个注释,为BEAST创建分区47.在检查每个位点中有多少多态位点和总位点后,我们决定使用四个分区:(1)属于蛋白质编码位置1和2的位点,(2)编码位置3,(3)RNA,或(4)非编码和非RNA。为了确保这些是高置信度的位点,每个分区也只包括那些在古序列中深度至少为20且在多序列对齐中总间隙小于3个的位置。这些分区分别有11,668、5,828、2,690和29,538个站点。我们使用这四个分区来运行BEAST47V1.10.4,为每个分区提供了非链接的替代模型和严格的时钟,每个分区具有不同的相对速率。(这些数据中没有足够的信息从单一校准中推断谱系间的比率变化)。我们将所有参考序列的年龄赋值为0,并对根高采用均值为61.1 Myr,标准差为1.633 Myr的正态分布先验48;通过保守地将95% HPD转换为得到标准偏差z分数。对于整体树先验,我们选择了合并模型。古序列的年龄是按照Shapiro et al.(2011)的总体程序估计的。98.为了评估对这个未知日期的先验选择的敏感性,我们使用了两种不同的先验,即gamma分布度量对更年轻的年龄(形状= 1,刻度= 1.7);以及在范围(0,10 Myr)上的均匀先验。我们还比较了每个分区内不同站点和替代类型之间的速率变化的两种不同模型,即由数据估计的四种速率类别的GTR+G模型和更简单的Jukes Cantor模型,该模型假设每个分区内替代类型和站点之间没有变化。所有其他先验都设置为默认值。无论是率模型还是先验选择都没有对结果产生定性影响(扩展数据图。10).我们还单独运行编码区域,因为它们翻译正确,因此是高度可靠的站点,并发现当使用较少的站点时,它们给出了相同的中位数和更大的置信区间(扩展数据图)。10).我们运行每个马尔可夫链蒙特卡洛,总共1亿次迭代。在去除前10%的老化后,我们验证了Tracer的收敛性91v1.7.2(迹的明显平稳性,所有参数的有效样本量为> 100)。我们还验证了TreeAnnotator生成的MCC树47将这个古老的序列在系统发育上与pathPhynder62如图扩展数据图所示。9.对于我们的主要结果,我们报告了统一的古代年龄先验,和GTR+G4模型应用到四个分区中的每一个。相关的XML在源数据3中给出。古代的95% HPD为(2.0172,0.6786)桦木属叶绿体序列,中位数估计为1.323 Myr,如图所示。2.
报告总结
有关研究设计的进一步资料,请参阅自然组合报告摘要链接到这篇文章。
数据可用性
原始序列数据(适配器修剪后的13135,646,556个读取)可通过ENA项目登录获得PRJEB55522.花粉计数可通过https://github.com/miwipe/KapCopenhagen.git.本文提供了原始数据。
代码的可用性
所有使用的代码都可以在https://github.com/miwipe/KapCopenhagen.git.
参考文献
萨尔兹曼等人。冰川变化和气候趋势来自秘鲁安第斯山脉南部数据稀缺的科迪勒拉·维尔卡诺塔地区的多个来源。冰冻圈7, 103-118(2013)。
联合国政府间气候变化专门委员会气候变化2013:自然科学基础(编斯托克,t.f.等人)(剑桥大学出版社,2013)。
Brigham-Grette, J.等人。在俄罗斯北极东北部记录的上新世变暖、极性放大和阶梯式更新世变冷。科学340, 1421-1427(2013)。
Gosse, J. C.等。PoLAR-FIT:上新世景观和北极遗迹-时间冻结。Geosci。可以。44, 47-54(2017)。
马修斯,j.v., Telka, A. Jr & Kuzmina, S. A.来自阿拉斯加和北极/亚北极加拿大的晚第三纪昆虫和其他无脊椎动物化石。黑旋风。Bespozvon。16, 126-153(2019)。
威勒斯列夫,E.等。全新世和更新世沉积物中多样的动植物遗传记录。科学300, 791-795(2003)。
Funder, S.等。上新世晚期格陵兰——格陵兰北部的Kap København地层。公牛。青烟。Soc。窝。48, 117-134(2001)。
范德尔,S. & Hjort, C.北格陵兰岛东部第四纪地质的侦察。拉普。Grønlands青烟。在20。99, 99-105(1980)。
Fredskild, B. & Røen, U.格陵兰北部Kap København间冰期泥炭沉积物中的大化石。北风之神11, 181-185(2008)。
Bennike, O. & Böcher, J.北极附近的森林苔原:格陵兰北部上新世-更新世Kap København组的植物和昆虫遗骸。北极43, 331-338(1990)。
德国人,J。格陵兰岛北部皮尔里地皮洛-更新世序列Kap Kobenhavn组的古昆虫学(Tusculanum博物馆出版社,1995年)。
Rybczynski, N.等。北极地区上新世中期的暖期沉积物使我们得以深入了解骆驼的进化。Commun Nat。4, 1550(2013)。
王X, Rybczynski, N., Harington, c.r ., White, s.c . & Tedford, r.h . A基熊(Protarctos abstrusus)揭示了欧亚的亲缘关系和富含可发酵糖的饮食。Sci代表。7, 17722(2017)。
西蒙纳森,洛杉矶,彼得森,K. S. &范德尔,S。格陵兰岛北部上新世-更新世Kap København组软体动物古生物学.第36卷(哥本哈根大学Tusculanum博物馆出版社,1998年)。
Mogensen, G. S.格陵兰岛北部Kap København的上新世或早更新世苔藓。Lindbergia10, 19-26(1984)。
Funder, S., Abrahamsen, N., Bennike, O. & feling - hanssen, R. W.北极森林:来自北格陵兰岛的证据。地质13, 542-546(1985)。
Abrahamsen, N. & Marcussen, C.北格陵兰岛东部上新世-更新世Kap København组的磁地层。理论物理。地球的星球。国际米兰。44, 53-61(1986)。
Bennike, O。Kap København组:格陵兰岛北部滩地上新世-更新世层序的地层和古植物学。Meddelelser om Grønland,地球科学Vol. 23 (Videnskabelige Umdersøgelser i Grønland的Kommissionen, 1990)。
费林-汉森,r.w.。格陵兰北部上新世-更新世Kap Kobenhavn组的有孔虫地层(Tusculanum博物馆出版社,1990)。
DNA初级结构的不稳定性和衰变。自然362, 709-715(1993)。
范德瓦尔克,T.等人。数百万年前的DNA揭示了猛犸象的基因组历史。自然591, 265-269(2021)。
Klimaet i Grønland。https://www.dmi.dk/klima/temaforside-klimaet-frem-til-i-dag/klimaet-i-groenland/(DMI, 2021)。
Allentoft, m.e.等人。DNA在骨中的半衰期:测定158个年代化石的衰变动力学。Proc,杂志。科学。279, 4724-4733(2012)。
质粒DNA在二氧化硅上的吸附:一价和二价盐的动力学和构象变化。《生物高分子8, 24-32(2007)。
梅尔扎克,K. A.舍伍德,C. S.特纳,R. F. B. &海恩斯,C. A.高氯酸盐溶液中二氧化硅吸附DNA的驱动力。胶体界面科学。181, 635-644(1996)。
蔡鹏,黄启勇。&张晓伟。DNA与粘土矿物和土壤胶体颗粒的相互作用及对DNA酶降解的保护作用。环绕。科学。抛光工艺。40, 2971-2976(2006)。
方勇,何俊华。亚精胺诱导云母表面DNA凝结的早期中间体。j。化学。Soc。120, 8903-8909(1998)。
卡尔博士& Bailiff博士水生环境中溶解核酸的测量和分布。Limnol。Oceanogr。34, 543-558(1989)。
Pedersen, m.w.等。北美无冰走廊的冰后生存能力和殖民。自然537, 45-49(2016)。
Meyer, M. & Kircher, M. Illumina测序库制备高复用目标捕获和测序。冷泉港。Protoc。2010db。prot5448(2010)。
默奇,t.j.等人。利用古芯片Arctic-1.0诱饵集优化古代环境DNA的提取和靶向捕获,以重建过去的环境。皮疹。Res。99, 305-328(2021)。
Benoit, G.等人。多重比较宏基因组学使用多集k- m计数。预印在https://arxiv.org/abs/1604.02412(2016)。
Pedersen, m.w.等。晚更新世黑熊和巨型短面熊的环境基因组学研究。咕咕叫。医学杂志。31, 2728 - 2736。e8(2021)。
王毅,等。从古代环境基因组学研究北极生物群晚第四纪动力学。自然600, 86-92(2021)。
Wang, Y. et al. ngslca -一个快速灵活的最低共同祖先推断和宏基因组数据分类分析的工具包。生态方法。另一个星球。https://doi.org/10.1111/2041-210X.14006(2022)。
Michelsen, C.等人metaDMG:一个古老的DNA损伤工具包。Zenodohttps://doi.org/10.5281/zenodo.7284374(2022)。
雷诺兹,m.k.等。一个栅格版本的北极圈植被图(CAVM)。遥感,环境。232, 111297(2019)。
格陵兰岛极地沙漠地带的植物区系和生态特征。j .蔬菜。科学。8, 685-696(1997)。
Boertmann, D. & Bay, C。Grønlands Rødliste 2018: Grønlandske Dyr og Planters Trusselstatus上的Fortegnelse(Grønlands自然研究所,奥胡斯大学,2018)。
Böcher, T. W.,霍尔曼,K. &雅各布森,K.;Grønlands植物第三版(Forlaget Haase & Søn, 1978)。
艾文,R.,默里,D. F.,拉兹文,V. Y. &尤尔采夫,B. A.。泛北极植物区系注释清单(空军)。http://panarcticflora.org(2022)。
格陵兰岛四十年来新的维管植物记录。PhytoKeys145, 63-92(2020)。
湾,C。北纬74度以北格陵兰岛北部维管植物的植物地理研究第36卷(Videnskabelige Undersøgelser i grnland的Kommissionen, 1992)。
Parducci, L.等。湖泊沉积物中的古代植物DNA。新植醇。214, 924-942(2017)。
也索斯,i.g.等。湖泊沉积物的植物DNA元条形码:它如何代表当代植被。《公共科学图书馆•综合》13, e0195403(2018)。
Bennike, O。Kap København组:格陵兰北部滩地上新世-更新世层序的地层和古植物学(Videnskabelige Undersøgelser i grnland的Kommissionen, 1990)。
德拉蒙德,A. J.和兰姆波特,A.野兽:贝叶斯进化分析的抽样树。BMC另一个星球。医学杂志。7, 214(2007)。
杨,X.-Y。et al。桦木科的质体及其系统发育意义。j .系统。另一个星球。57, 508-518(2019)。
Bouckaert等人。BEAST 2:贝叶斯进化分析软件平台。公共科学图书馆第一版。医学杂志。10, e1003537(2014)。
多塞特,H. J.,钱德勒,M. A.,克罗宁,T. M. & Dwyer, G. S.上新世中期海面温度变化。古海洋学https://doi.org/10.1029/2005PA001133(2005)。
格雷厄姆,R. W.等。阿拉斯加圣保罗岛全新世中期猛犸象灭绝的时间和原因。国家科学院学报美国113, 9310-9314(2016)。
Parducci, L.等。鸟枪环境DNA,花粉,和大化石分析的湖区沉积物从瑞典南部。前面。生态。另一个星球。7, 189(2019)。
Rijal, D. P.等。沉积古DNA显示Fennoscandia北部全新世以来陆生植物丰富度不断增加。科学。睡觉。7, eabf9557(2021)。
Cocker, s.l.等。东米尔福德乳齿象的粪便分析:新斯科舍省中部75 ka年的饮食和环境重建英国石油公司.可以。地球科学。https://doi.org/10.1139/cjes-2020-0164(2021)。
弗莱彻,T. L., Telka, A., Rybczynski, N. & Matthews, J. V. Jr.美国阿拉斯加和北极/亚北极加拿大的新近纪和早期更新世植物群:新数据,洲际比较和相关性。Palaeontol。电子乐https://doi.org/10.26879/1121(2021)。
冯,R.等。由于更大的辐射强迫和关闭的北冰洋门户,北部高纬度地区的上新世晚期陆地温暖被放大。地球的星球。科学。列托人。466, 129-138(2017)。
加莱蒂等人。巨型动物灭绝的生态和进化遗产。医学杂志。坎布·菲洛斯牧师。Soc。93, 845-862(2018)。
Malhi, Y.等人。从更新世到人类世的巨型动物和生态系统功能。国家科学院学报美国113, 838-846(2016)。
罗斯塔德,J.,布兰克,Y.-L。芬诺斯堪地北部森林西部的火灾历史受人类土地利用和气候的影响。生态。Monogr。87, 219-245(2017)。
Elias, S. A. & Matthews, J. V. Jr北极北美从中新世晚期到更新世早期的季节温度,基于化石甲虫组合的相互气候范围分析。可以。地球科学。39, 911-920(2002)。
格陵兰岛东北部第四纪一个显著的有孔虫组合。公牛。青烟。Soc。丹麦38, 101-107(1989)。
martininiano, R., De Sanctis, B., Hallast, P. & Durbin, R.将古代DNA序列放入参考系统发育中。摩尔。杂志。另一个星球。39, msac017(2022)。
德尔蒙特,T. O.等人。在阳光照射的海洋中丰富的远亲真核浮游生物谱系的功能库收敛。细胞染色体组。2, 100123(2022)。
林达尔,T. &尼伯格,B.天然脱氧核糖核酸的去嘌呤率。生物化学11, 3610-3618(1972)。
汉森,J.,佐藤,M.,罗素,G.和哈雷查,P.气候敏感性,海平面和大气二氧化碳。费罗斯。反式。一个371, 20120294(2013)。
Taut, T., Kleeberg, R. & Bergmann, J.新的Seifert Rietveld程序BGMN及其在定量相分析中的应用。板牙。结构体。5, 57-66(1998)。
Doebelin, N. & Kleeberg, R. Profex: Rietveld精炼程序BGMN的图形用户界面。j:。Crystallogr。48, 1573-1580(2015)。
谢瑞,R. W. &科埃略,A. x射线线轮廓拟合的基本参数方法。j:。Crystallogr。25, 109-121(1992)。
凯斯特,D. R.,杜达尔,I. W.,康纳斯,D. N. &皮特科维茨,R. M.人工海水的制备1。Limnol。Oceanogr。12, 176-179(1967)。
格里丘克,E. D. &扎克林斯卡娅,V. P。花粉、孢子化石的分析及其古地理应用(地理出版社,1948)。
Kupriyanova,洛杉矶& Aleshina,洛杉矶。苏联欧洲区系的花粉和孢子(Nauka, 1972)。
摩尔,P. D.韦伯,J. A.和柯林森,M. E.。花粉分析.(Blackwell Scientific, 1991)。
格林,e.c。Tilia和Tiliagraph(伊利诺伊州立博物馆,1991)。
本内特,K. D. psimpoll和pscomb手册。http://www.chrono.qub.ac.uk/psimpoll/psimpoll.html(2002)。
Ardelean, C. F.等。末次盛冰期人类在墨西哥居住的证据。自然584, 87-92(2020)。
Wood, D. E., Lu, J. & Langmead, B.改进的宏基因组分析与Kraken 2。基因组医学杂志。20., 257(2019)。
Karsenti, E.等人。海洋生态系统生物学的整体方法。公共科学图书馆杂志。9, e1001177(2011)。
朗米德,B. &萨尔茨伯格,S. L.快速间隙阅读对齐与领结2。Nat方法。9, 357-359(2012)。
Eren, a.m.等人。社区主导,集成,可重复的多组学与anvi 'o。Microbiol Nat。6, 3-6(2021)。
被子植物系统发育类群。一个nupdate of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG II.机器人。j·林恩。Soc。141, 399-436(2003)。
Chevigny, N., Schatz-Daas, D., Lotfi, F. & Gualberto, J. M. DNA修复和植物线粒体基因组的稳定性。Int。理学。21, 328(2020)。
克拉克,K., Karsch-Mizrachi, I., Lipman, d.j., Ostell, J. & Sayers, E. W. GenBank。核酸测定。44, d67-d72(2016)。
也索斯,i.g.等。末次冰期最盛期在挪威北部Andøya的环境条件;北部冰缘生态“热点”的证据。皮疹。科学。牧师。239, 106364(2020)。
Katoh, K. & Standley, D. M. MAFFT多序列比对软件版本7:性能和可用性的改进。摩尔。杂志。另一个星球。30., 772-780(2013)。
Yachdav, G.等人。MSAViewer:多个序列对齐的交互式JavaScript可视化。生物信息学32, 3501-3503(2016)。
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X:跨计算平台的分子进化遗传学分析。摩尔。杂志。另一个星球。35, 1547-1549(2018)。
Cock, P. J. A.等。BioPython:用于计算分子生物学和生物信息学的免费Python工具。生物信息学25, 1422-1423(2009)。
佩奇,A. J.等人。SNP-sites:从multi-FASTA比对中快速高效地提取snp。活细胞。染色体组。2, e000056(2016)。
李,H. & Durbin R.快速和准确的短读对齐与Burrows-Wheeler变换。生物信息学25, 1754-1760(2009)。
李,H.等。序列对齐/映射格式和SAMtools。生物信息学25, 2078-2079(2009)。
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A.使用示踪器1.7在贝叶斯系统发育中的后验总结。系统。医学杂志。67, 901-904(2018)。
Karpinski, E.等人。美洲乳齿象线粒体基因组表明,在更新世气候振荡的反应中,有多次分散事件。Commun Nat。11, 4048(2020)。
黄艳,王娟,杨艳,范晨,陈杰。水杨科植物叶绿体基因组的系统基因组学分析。前面。植物科学。8, 1050(2017)。
Fourment, M. & Holmes, E. C. Seqotron: Mac OS X的用户友好的序列编辑器。BMC Res. Notes9, 106(2016)。
李,H.等。Seqtk:用于处理FASTA或FASTQ序列的快速轻量级工具。https://github.com/lh3/seqtk(2018)。
通过碱基比对质量提高SNP发现。生物信息学27, 1157-1158(2011)。
R核心团队。R:用于统计计算的语言和环境。R统计计算基金会,维也纳,奥地利。https://www.R-project.org/(2018)。
夏皮罗等人。一种估计未知序列年龄的贝叶斯系统发育方法。摩尔。杂志。另一个星球。28, 879-887(2011)。
黄,D. I., Hefer, C. A., Kolosova, N., Douglas, C. J. & Cronk, Q. C. B.全质体测序揭示了近亲缘香脂杨树之间的深层质体分化和细胞核不一致。杨树balsamifera而且p . trichocarpa(杨柳科)。新植醇。204, 693-703(2014)。
李文华,李文华,李文华。更新世植物的物种形成杨树(杨柳科)。系统。医学杂志。61, 401-412(2012)。
张丽丽,奚忠,王明,郭晓霞,马涛。水杨科植物质体系统发育与谱系多样化研究——以杨树和柳树为例。生态。另一个星球。8, 7817-7823(2018)。
确认
我们感谢嘉士伯基金会在2006年和2012年对Kap København进行两次考察的后勤支持(S. Funder,嘉士伯基金会资助LongTerm和Kap København - the age的首席研究员)。2016年的实地调查得到了维卢姆基金会(Villum Foundation)对N.K.L.的资助。我们非常感谢Illumina公司的合作支持,这对项目的成功至关重要。E.W.和K.H.K.感谢丹麦国家研究基金会(DNRF)和Lundbeck基金会(r202 -2018-2155)提供长期资金,开发必要的DNA技术,最终使从Kap København组的这些古老沉积物中提取环境DNA成为可能。E.W.还感谢惠康基金会(UNS69906)、嘉士伯基金会(CF18-0024)、Novo基金会(NNF18SA0035006)、Leverhume (pg -2016-235)和GRF EXC CRS主席-卓越集群(44113220)的支持。M.W.P.感谢嘉士伯基金会(CF17-0275)的支持。K.K.S.和S.J.感谢VILLUM FONDEN(00025352)的支持。根据欧盟地平线2020研究和创新计划,I.G.A.和E.C.获得了欧洲研究理事会(ERC)的资助(资助协议编号:no. 1)。819192)。B.D.S.感谢威康基金会数学基因组学和医学项目(WT220023)的支持。 J.Å.K. was supported by the Carlsberg Foundation (CF20-0238). C.B. acknowledges ERC Advanced Award Diatomic (grant agreement no. 835067). J.C.G. was supported by Natural Science and Engineering Research Council of Canada–Discovery Grant 06785 and Canada Foundation for Innovation grant 21305. M.J.C. acknowledges support from the Danish National Research Foundation DNRF128. We thank G. Yang for cosmogenic isotope AMS target chemistry; S. Funder for introducing us to the Kap København Formation and generating much of the platform that enabled us to conduct our research; T. O. Delmont for providing data and guidance on the SMAGs analysis; Minik Rosing for providing talc minerals; T. B. Zunic for providing tremolite, orthoclase and chlorite; Z. Vardanyan for help with the DNA extractions and library build; and L. B. Levy and D. Skov for their help collecting samples in 2016. This work was prepared in part by Lawrence Livermore National Laboratory under contract DE-AC52-07NA27344; LLNL-JRNL-830653. E.W. thanks St Johns College, Cambridge for providing him with a stimulating environment for scientific thoughts and discussion.
作者信息
作者及隶属关系
财团
贡献
K.H.K.和E.W.构想了这个想法。k.h.k.、M.W.P.和E.W.设计了这项研究。k.h.k.、a.m.a.s.、a.s.t.、N.K.L.和E.W.提供了样本、背景并进行了实地调查。M.W.P.进行了DNA实验室分析和分类学分析。M.W.P.、B.D.S.和B.D.C.在M.S.和R.D.的监督下进行了系统发育定位。B.D.S.、M.W.P.和B.D.C.在R.D.和J.J.W.的监督下进行了基因年代测定。M.W.P.、T.S.K.和C.S.M.构思、设计并进行了DNA损伤估计。K.K.S.和S.J.构思并设计了这项研究的dna -矿物方面,解释并撰写了dna -矿物数据,并参与了热年龄计算。k.h.k.、M.W.P、a.h.r.、a.r.、I.G.A.和E.W.进行了区系分析和解释。K.K.K.进行制图和GIS分析。I.S.设计并进行了古地磁分析,并解释了结果。J.C.G.准备并分析了8个宇宙成因样品26艾尔和10贝和a。j。h。解释了他们的埋葬年龄。i.g.a.、E.C.和Y.W.提供了对PhyloNorway参考数据库的访问,并为叶绿体的系统发育位置提供了输入。a.s.t检测了另外六个样本的花粉。J.A.K.支持沉积物物源评价。M.B.提供了北格陵兰岛的矿物学数据。c.d., m.r., M.E.J.和B.S.设计并实施了基于ddPCR的检测和鉴定样品中古代植物DNA的方法。A.F.-G。对SMAGs的生物信息学分析有贡献,C.B.对海洋宏基因组信号的解释有贡献。M.J.C.对热年龄和DNA模型做出了贡献。 M.E.A. contributed to the DNA decay rate estimates. K.H.K., M.W.P., A.H.R. and E.W. interpreted the results and wrote the manuscript with contributions from K.K.S., S.J., A.R., B.D.S., B.D.C., I.G.A., J.C.G., I.S. and N.K.L., with inputs from the other authors.
相应的作者
道德声明
相互竞争的利益
作者声明没有利益竞争。
同行评审
同行评审信息
自然感谢匿名审稿人对本工作的同行评审所作的贡献。
额外的信息
出版商的注意施普林格自然对出版的地图和机构从属关系中的管辖权主张保持中立。
扩展的数据图形和表格
图1扩展数据
在队形中键入50个指示单位b.带完整沉积序列的74a+b区概况。C.局部性概述D.挖掘和清理古eDNA样本前B3单元富含有机沉积物的细节。E.在50号地点B3单元的永久冻土带取样。F.74a+b地块B2单元大型交错层理底部的富有机质沉积物。白色的圆圈表示人的比例。
图2 Leporidae线粒体reads的系统发育放置结果,仅使用转位snp。
读取已从所有层和站点合并。每个边缘上的绿色数字表示支持(+)snp的数量,而红色数字表示古Leporidae环境线粒体基因组中冲突的(−)snp与分配到各自边缘的参考snp重叠。有一个明确的位置为古Leporidae环境线粒体基因组的边缘标记+2,以现存的基础天兔座血统。
扩展数据图3 Capreolinae线粒体reads的系统发育放置结果,仅使用转位snp。
读取已从所有层和站点合并。每条边的绿色数字表示支持(+)snp的数量,而红色数字表示在古Capreolinae环境线粒体基因组中冲突的(−)snp与分配到各自边缘的参考snp重叠。有一个明确的位置为古羊角菜科环境线粒体基因组的边缘标记为+8/−3,基部为至今属。
图4象皮科线粒体reads在乳齿象内的系统发育位置(Mammut americanum),使用Elephas马克西姆斯为外群,包括转换和转位snp。
(请注意,NCBI分类法包括象皮科中的猛犸象属)。参考数据集包括乳齿象线粒体(Mammut americanum)只有一个Elephas马克西姆斯作为一个外部团体。读取已从所有层和站点合并。每条边的绿色数字表示支持(+)snp的数量,而红色数字表示古象皮科环境线粒体基因组中冲突的(−)snp与分配到各自边缘的参考snp重叠。边缘上有一个标记为+2/−1的古象科环境线粒体基因组的位置,表明它位于乳齿象的基底(Mammut americanum)演化支,其中包含大部分乳齿象参考线粒体基因组。请注意,这个位置是基于两个转换SNP,每个SNP的读取深度为3个读取。
扩展数据图5仅使用转位snp对鹅科中指定的线粒体reads和鹅科的代表进行系统发育放置。
读取已从所有层和站点合并。每个边缘上的绿色数字表示支持(+)snp的数量,而红色数字表示古鹅科环境线粒体基因组中冲突的(−)snp与分配到各自边缘的参考snp重叠。有一个明确的位置为古雁科环境线粒体基因组的边缘标记+3,基向Branta属。
图6蟋蟀科线粒体reads的系统发育放置结果,仅使用转位snp。
读取已从所有层和站点合并。每条边的绿色数字表示支持(+)snp的数量,而红圈中的红色数字表示古蟋蟀科环境线粒体基因组中冲突的(−)snp与分配到各自边缘的参考snp重叠。在边缘上有一个标记为+1的古蟋蟀科环境线粒体基因组的位置,是Arvicolinae亚科的基础。
扩展数据图7我们的系统发育放置结果杨树叶绿体读取,使用转换和反转snp,并使用从所有层和位点合并的读取。
每条边的数字代表古代支持(+)和冲突(−)snp的数量杨树环境基因组重叠的参考snp分配到边缘。古代的杨树环境基因组显然包含了不同物种的混合物。最可能的位置是上面的边缘杨树trichocarpa(NC 009143.1)和杨树balsamifera(NC 024735.1), +71/−15支持和冲突的snp。然而,我们发现直接导致这些物种的两个分支也有一些支持。杨树balsamifera而且p . trichocarpa被认为是姐妹物种。它们都分布在北美,北至阿拉斯加,以杂交而闻名杨树物种和形态非常相似93,99,One hundred..之前的分析发现,最近的核基因组分化时间仅为75000年前杨树trichocarpa而且p . balsamiferaOne hundred.,但深层叶绿体基因组分化时间至少为6-7 Ma99这在植物中并不常见。我们的古代杨树样本可以包含现代人的祖先或杂交的个体杨树trichocarpa而且p . balsamifera物种。
扩展数据图8我们的系统发育位置结果柳树叶绿体读取,使用转换和反转snp,并使用从所有层和位点合并的读取。
每条边的数字代表古代支持(+)和冲突(−)snp的数量柳树环境基因组重叠的参考snp分配到边缘。古代的柳树环境基因组是一个主要的基础基因组柳树进化枝。我们的古代柳树样本在系统发育上被放置在通往主分支的基枝上,有356个支持snp和22个冲突snp。虽然柳树叶绿体系统发育还没有完全解决93,分辨率的困难在于我们的放置分支,这与放置分支上的大量snp一起,使我们对放置位置有信心。我们的叶绿体系统发育大致符合101,估计了这两个主要指标之间的分歧日期柳树第一个演化支的根龄为8.1 Ma,我们的古样品是基于这个演化支的。因此,我们有理由得出结论,我们的古代柳树样品与现代样品至少相差8.1 Ma柳树物种,并可能代表一个灭绝的物种,或没有参考基因组测序的现存物种,或它们的集合。
扩展数据图9我们的系统发育放置结果桦木属叶绿体读取,使用转换和反转snp,并使用从所有层和位点合并的读取。
我们的古代桦木属样品放在主管的基底上桦木属该分支基于其定位分支上的29个支持(绿色)和13个冲突(红色)snp,并且除了导致该分支的snp外,树中其他地方的支持snp数量非常少。该位置与BEAST分子测年分析(见分子测年方法)一致。
图10分子年龄分布。
结果跑了古桦木属叶绿体分子年代测定分析47)与不同的先验和核苷酸取代模型。只使用编码区域,因此使用较少的总站点,可以得到预期的更大的置信区间。文中报告的结果为红色曲线,具有平坦先验和GTR+Γ4替换模型。
补充信息
补充信息
该文件包含背景,以及额外的信息和讨论的方法和中间结果有关的主要发现古地磁,以前的年龄控制,宇宙成因定年,矿物学,微观和宏观化石分析,并在主要文本中提出的eDNA。它还列出了PhyloNorway财团成员。
补充数据1
该文件包含了DNA序列元数据和样本地质单元信息的表格,DNA、大化石、系统发育位置之间的比较,为基因组数据库添加的附加参考,数字液滴PCR结果,以及用于绘制分类剖面的表格(包括原始计数和百分比)。
补充数据2
补充数据2包含所有x射线衍射结果,矿物实验处理和吸附,以及热年龄计算。
补充数据3
补充数据3是带有。xml文件的桦木属分子年龄估计。
补充数据4
获得的单个x射线衍射剖面。
补充数据5
补充数据5为计算出的强度及其差异,格式为.tiff文件。
权利和权限
开放获取本文遵循知识共享署名4.0国际许可协议(Creative Commons Attribution 4.0 International License),允许以任何媒介或格式使用、分享、改编、分发和复制,只要您对原作者和来源给予适当的署名,提供知识共享许可协议的链接,并注明是否有更改。本文中的图像或其他第三方材料包含在文章的创作共用许可中,除非在材料的信用额度中另有说明。如果内容未包含在文章的创作共用许可协议中,并且您的预期使用不被法定法规所允许或超出了允许的使用范围,您将需要直接获得版权所有者的许可。要查看此许可证的副本,请访问http://creativecommons.org/licenses/by/4.0/.
关于本文

引用本文
kæ r, k.h., Winther Pedersen, M., De Sanctis, B.。et al。环境DNA发现了格陵兰岛200万年的生态系统。自然612, 283-291(2022)。https://doi.org/10.1038/s41586-022-05453-y
收到了:
接受:
发表:
发行日期:
DOI:https://doi.org/10.1038/s41586-022-05453-y
这篇文章被引用
快速进化的基因组区域是人类大脑发育的基础
自然(2023)
Walker & Syers模型和土壤年龄
土壤生态学通讯(2023)
最古老的DNA显示乳齿象在200万年前漫步在格陵兰岛
自然(2022)
